

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Acromegaly is a condition caused by elevated levels of growth hormone (GH) over a prolonged period.1
Acromegaly occurs post-epiphyseal fusion. In contrast, gigantism is a condition of childhood due to excessive growth hormone secretion prior to epiphyseal growth plate fusion.2
It is normally a sporadic condition seen in middle-aged adults with equal distribution between males and females. However, rarer familial causes can present in younger patients.2
Familial causes of acromegaly include multiple endocrine neoplasia type 1 (MEN-1), McCune-Albright syndrome and Carney complex.2
Aetiology
Acromegaly is usually caused by a growth hormone (GH)-secreting pituitary adenoma. Rarely, pituitary hyperplasia may also result in elevated GH.1,3
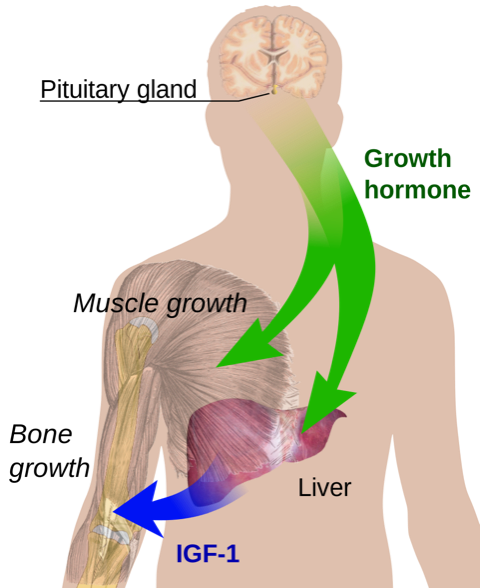
IGF-1 is produced in response to GH. IGF-1 mediates the effects of GH and has a negative feedback effect to inhibit GH secretion by producing somatostatin and acting on somatotrophs (Figure 1).3
For more information, see the Geeky Medics guide to the pituitary gland.
In the case of acromegaly, the negative feedback loop is broken leading to increasing levels of GH.
Clinical features
History
Typical symptoms of acromegaly include:1,4
- Symptoms due to tumour: headache, visual changes
- Systemic symptoms: change in appearance, weight gain, fatigue, snoring, joint pain, voice changes, skin tags
- Symptoms due to prolactin: amenorrhoea, galactorrhoea, erectile dysfunction, reduced libido, infertility
Other important areas to cover in the history include:
- Family history: multiple endocrine neoplasia type 1 (MEN-1), McCune-Albright syndrome and Carney complex.
Clinical examination
Typical clinical findings in acromegaly include:1,4
- Changes in appearance: frontal bossing (prominent forehead), macroglossia (large tongue), prognathism (protruding lower jaw), interdental separation, enlarged hands & feet, skin thickening
- Effects on hands: tight rings on fingers, signs of carpal tunnel syndrome
- Visual field defects: bitemporal hemianopia due to pituitary adenoma
- Other signs: galactorrhoea, hypertension, signs of heart failure
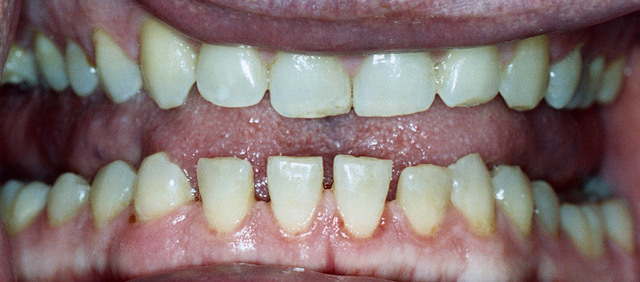
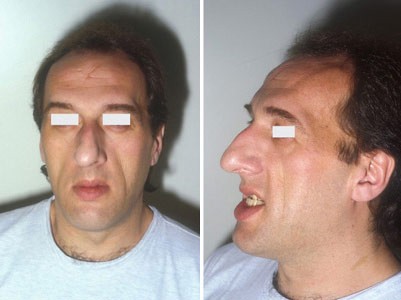
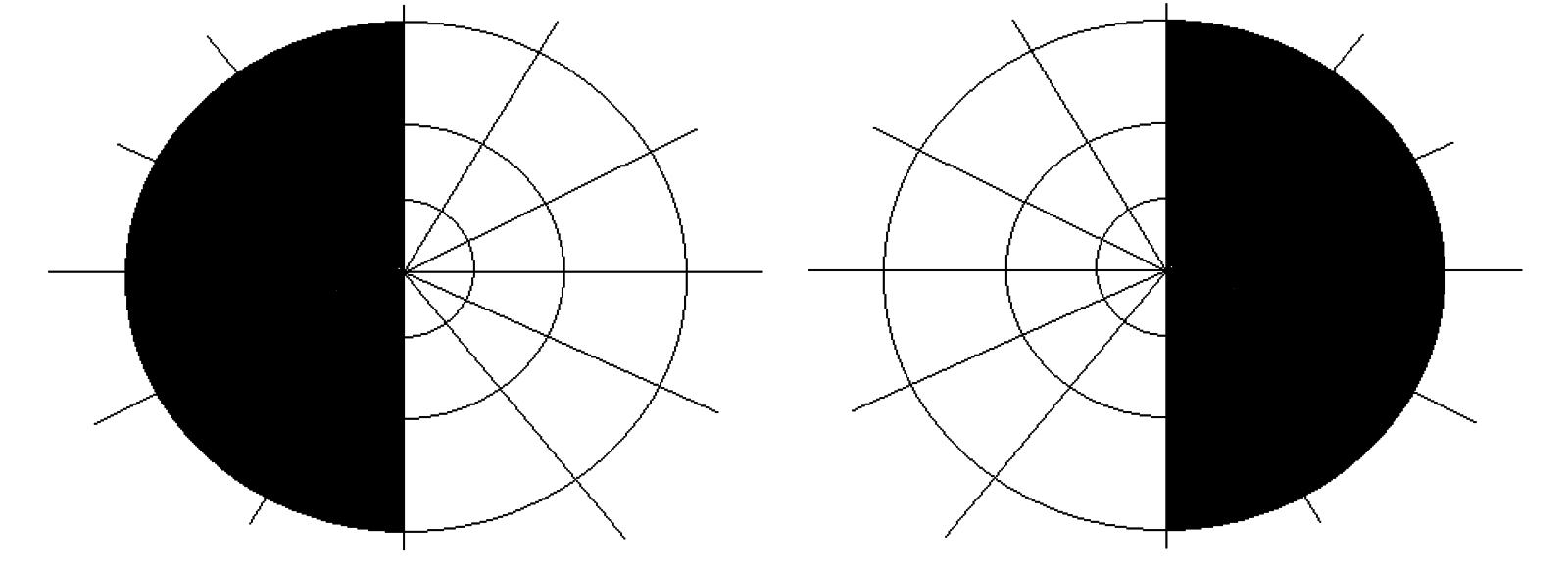
Investigations
Bedside investigations
Relevant bedside investigations include:2
- Visual field examination: a pituitary adenoma impacting the optic chiasm will cause bitemporal hemianopia
Laboratory investigations
Relevant laboratory investigations include:1,2
- IGF-1: this will be raised
- Oral glucose tolerance test (OGTT): this is used to confirm a diagnosis. Glucose normally suppresses GH. In acromegaly, GH will not be suppressed after an OGTT.
- Blood glucose, calcium, phosphate and triglycerides: these may be raised in acromegaly.
- Pituitary assessment (prolactin, cortisol, thyroid function tests, FSH/LH, oestradiol and testosterone): to assess pituitary function.
Serum growth hormone is not routinely used due to GH pulses throughout the day and at times of stress
Imaging
Relevant imaging for acromegaly include:2
- MRI pituitary: to investigate for a pituitary adenoma
Management
The goal of management is to normalise hormone levels and remove the pituitary adenoma (if possible).
Surgical management
Surgical removal of the pituitary adenoma is usually performed via transsphenoidal surgery. This is the initial treatment for most patients and involves making an incision in the nose (sphenoid sinus) to allow access to the pituitary adenoma.6
Patients can be considered cured after surgery but will need regular follow-up.5
Medical management
Medical management is normally used as adjuvant treatment or for patients where surgery is not recommended.3
Somatostatin analogues
Somatostatin analogues inhibit the release of growth hormone. Examples include octreotide and lanreotide.2
These medications do not cure acromegaly, but they have been shown to reduce tumour size.3
Gastrointestinal side effects are common and include symptoms such as cramping, bloating and diarrhoea. These symptoms occur initially after starting treatment but usually resolve once established on the medication. Up to 20% of patients can develop gallstones.6
Growth hormone receptor antagonists
Pegvisomant inhibits the synthesis of IGF-1 by acting as a growth hormone receptor antagonist.2 It is used as a second-line option if somatostatin analogues fail to control symptoms or hormone levels.3
Growth hormone antagonists can cause abnormalities in liver function. This is usually reversible and liver function tests should be monitored.6
Dopamine agonists
Dopamine agonists (e.g. bromocriptine and cabergoline) work by binding to dopamine receptors in the pituitary which suppresses GH secretion. They also suppress prolactin secretion.3
Side effects of dopamine agonists include nausea, fatigue, dizziness due to postural hypotension and nightmares.6
Radiotherapy
Pituitary radiotherapy (most often, stereotactic gamma-knife) is used when there is a failure of surgical or medical treatment and can be used in combination with medical therapy.3
Radiotherapy carries the risk of hypopituitarism which can affect fertility.1
Long term monitoring
It’s important to follow up patients to monitor disease activity and complications that can result from acromegaly.
Disease activity monitoring includes:6
- IGF-1
- Change in signs and symptoms
- Pituitary hormone tests: TFT, cortisol, prolactin, oestradiol and testosterone
- MRI pituitary: to monitor the size of the adenoma.
Monitoring for complications may include:3
- Blood pressure measurements
- ECG and echocardiography
- Epworth sleepiness scale
- OGTT
- Colonoscopy every 10 years (due to the increased risk of colonic polyps)
Complications
Cardiovascular disease (including hypertension, congestive cardiac failure, stroke and coronary heart disease) is a major complication of acromegaly.2
Other complications include:2
- Diabetes mellitus
- Obstructive sleep apnoea
- Colorectal cancer: acromegaly increases the risk of colonic polyps.
- Thyroid cancer
- Hypopituitarism
- Carpal tunnel syndrome
- Osteoarthritis
- Psychosocial impacts
Key points
- Acromegaly is a rare endocrine condition due to elevated GH levels
- The most common cause is a pituitary adenoma
- Typical symptoms of acromegaly include headache, change in appearance, weight gain, vision changes and daytime tiredness
- Typical clinical signs of acromegaly include bitemporal hemianopia, prominent forehead, interdental separation, prognathism and large tongue
- Relevant investigations in suspected acromegaly include serum IGF-1 levels and an oral glucose tolerance test
- Definitive treatment of acromegaly involves the removal of the pituitary adenoma via transsphenoidal surgery.
- The most common complications of acromegaly are cardiovascular disease. Patients are at an increased risk of developing thyroid or colorectal cancer.
Reviewer
Dr Michael Phipps
Undergraduate Tutor
Editor
Dr Chris Jefferies
References
- Kumar P, Clark M. Kumar & Clark clinical medicine. 9th ed. Edinburgh: Elsevier Saunders; 2017.
- Payne, J., 2016. Acromegaly | Doctor’s Guide. [online] Patient.info. Available from: [LINK]
- Dineen R, Stewart P, Sherlock M. Acromegaly – Diagnosis and Clinical Management. QJM [Internet]. 2016. Available from: [LINK]
- Adigun, O., Nguyen, M., Fox, T. and Anastasopoulou, C., 2022. Acromegaly. [online] Ncbi.nlm.nih.gov. Available from: [LINK]
- Behari S, Banerji D, Das N, Sharma S, Jindal Y, Jain V. Surgical management of acromegaly: Long term functional outcome analysis and assessment of recurrent/residual disease. Asian Journal of Neurosurgery [Internet]. 2016 ;11(3):261. Available from: [LINK]
- Melmed S, Katznelson L. UpToDate [Internet]. Uptodate.com. 2022. Available from: [LINK]
Image references
- Figure 1. Mikael Haggstrom. Endocrine growth regulation. Licence: [Public domain]
- Figure 2. Offices of Kenneth Yamanaka. Acromegaly teeth gapping. Licence: [Public domain]
- Figure 3. Philippe Chanson and Sylvie Salenave. Prognathism, maxillary widening, teeth separation and jaw malocclusion. Licence: [CC BY 2.0]
- Figure 4. RobertB3009. Bitemportal hemianopia. Licence: [CC BY-SA 4.0]
