

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Behcet’s disease is presumed to be an autoimmune vasculitis, though its cause is unknown. It is characterised by oral and genital ulcer formation with uveitis.
Behcet’s disease is most prevalent in Mediterranean, Middle Eastern, and East Asian populations. Turkey has the highest prevalence at 420 cases per 100,000 population. In the United States, the prevalence is much lower at roughly 5 per 100000 population.1,2
Aetiology
The aetiology of Behcet’s disease is currently unknown.
Epidemiological studies suggest that there may be a genetic basis to the development of Behcet’s disease, as those with HLA-B51/B5 carriers are most likely to develop the condition. However, the inheritance of Behcet’s disease is non-Mendelian.2
One current theory is that exposure to Staphylococcus aureus or to other pathogenic antigens may serve as a trigger for the development of the autoimmune response.2
Risk factors
Risk factors for Behcet’s disease include:1,2
- Mediterranean, Middle Eastern, and East Asian ethnicity
- Age 20–30 years
- HLA-B51 carrier
Disease prevalence is roughly equal among males and females but males tend to have more severe presentations.
Clinical features
The classic triad of the disease is oral ulcers, genital ulcers, and uveitis.
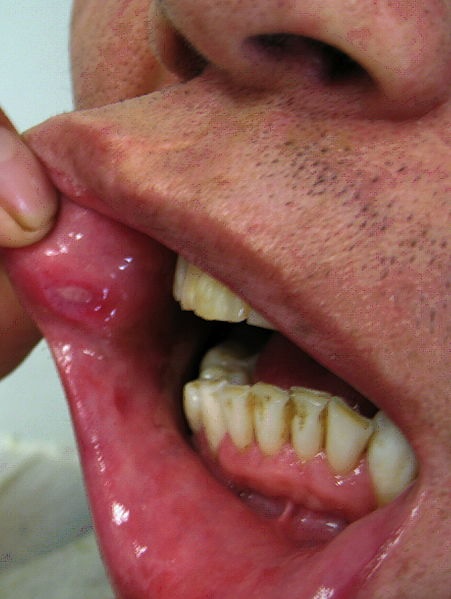
Multiple painful ulcers of the oral mucosa are the hallmark of Behcet’s disease. These can present anywhere in the oral mucosa and are recurrent, often last for several weeks before healing.1
Genital ulcers often occur on the penis in males and are extremely painful. They may also last several weeks before resolving. However, unlike oral ulcers, these are more likely to scar.2
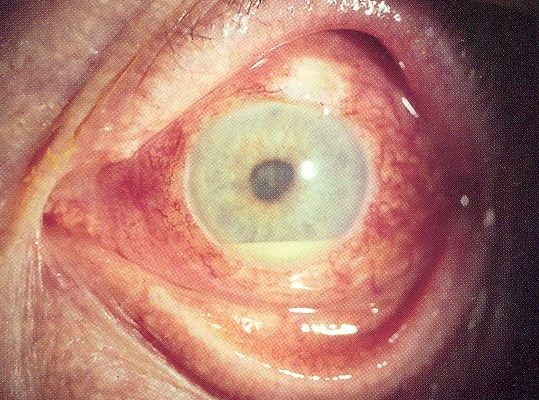
Uveitis may develop several years after the initial presentation of the disease. Patients may complain of bilateral vision loss.2
Other systemic manifestations include:1,2
- Memory impairment
- Loss of coordination
- Deep vein thrombosis
- Gastrointestinal ulceration
- Inflammatory, non-erosive arthritis (most commonly of the knees)
- Fever
- Rash
History
Typical symptoms of Behcet’s disease include:
- Painful ulcers in the mouth (Figure 1)
- Painful ulcers of the genitalia
- Blurred vision
- Joint pains
- Fever
Clinical examination
Typical clinical findings in Behcet’s disease include:2
- Ulcers of the mouth and genitalia
- Reduced visual acuity
- Erythema of the eyes (Figure 2)
- Erythema nodosum
Differential diagnoses
Several other diseases can mimic both the ulcers and the other systemic features seen in Behcet’s disease:1,2
- Crohn’s disease
- Herpes simplex virus
- Hand foot and mouth disease
- Systemic lupus erythematosus
- Seronegative arthritis
- Multiple sclerosis
- Takasyu’s arteritis
- Sarcoidosis
- Squamous cell carcinoma
Investigations
Most investigations in the work-up of Behcet’s disease are performed for the purposes of ruling out other diagnoses.
Bedside investigations
Relevant bedside investigations include:
- Pathergy testing: though this is less commonly positive in Caucasian patients.5
Pathergy test
The pathergy test is a hypersensitivity reaction to pinprick. A small needle is inserted into the skin and the needle site is evaluated at 24 and 48 hours.
A positive finding is the formation of a small papule at the insertion site, which is suggestive of Behcet’s syndrome.5
Laboratory investigations
Relevant laboratory investigations include:1
- Haematology: full blood count (may show anaemia of chronic disease and leukocytosis)
- Biochemistry: elevated inflammatory markers (ESR, CRP, complement, acute phase reactants)
- Immunology: antiphospholipid antibodies (may be positive in some cases)
- Genetics: HLA-B51 testing
Imaging
Relevant imaging investigations depend on clinical indications but may include:1
- X-rays of affected joints
- CT scan: if suspicious of bleeding, thrombosis, or other complications
- CT angiography: to evaluate for the presence of large vessel thrombosis or aneurysm
- MRI brain
Diagnosis
The diagnosis of Behcet’s disease is entirely clinical, and there are no definitive laboratory investigations to confirm the diagnosis.
The diagnostic criteria for Behcet’s disease are described in The International Clinical Criteria for Behcet’s Disease Classification. A diagnosis requires recurrent oral ulcers at least three times in the past year AND…
Any two of the following:6
- Recurrent genital ulcerations
- Uveitis or retinal vasculitis
- Skin lesions
- Positive pathergy test
Management
The management of Behcet’s disease is aimed at controlling symptoms and inflammation in the affected systems.7,8
Corticosteroids and colchicine are used as first-line agents in the treatment of oral and genital ulcers. Immunosuppressive agents (e.g. azathioprine, anti-tumour necrosis factor-a antibodies) should be used in select cases when the previously mentioned drugs are unsuccessful.
For patients with additional systemic involvement, corticosteroids and immunosuppressant medications are used to reduce inflammation and control symptoms. Patients with eye symptoms should be referred to ophthalmology.
Complications
Behcet’s disease with eye involvement increases the risk of permanent visual impairment and blindness.
Systemic involvement can increase the risk of central nervous system defects, cardiovascular complications, gastrointestinal haemorrhage, and thrombosis.2
Vascular inflammation can increase the risk of coronary and pulmonary aneurysms, which can rupture and lead to mortality.2
Prognosis
Prognosis is variable and can depend on many factors.
In some patients, systemic complications can lead to early mortality. Complications associated with a worse prognosis include ruptured aneurysms, renal amyloidosis, and neurological sequelae. Prognosis is worse in men and those diagnosed at a younger age.2
With proper medical management, most patients can live normal lives.6
Key points
- Behcet’s disease is a systemic autoimmune vasculitis with an unknown aetiology.
- Risk factors for Behcet’s disease include ethnicity (Mediterranean, Middle Eastern, and East Asian) age 20–30 years, and being an HLA-B51 carrier.
- The hallmark of Behcet’s disease is a triad of oral ulcers, genital ulcers, and uveitis.
- Other clinical features include gastrointestinal ulcers, deep vein thrombosis, and neurological sequelae.
- The pathergy test is a common test used in Behcet’s classification.
- The International Clinical Criteria for Behcet’s Disease Classification are used in the classification of Behcet’s disease.
- Corticosteroids and colchicine are the first-line agents used in the management of Behcet’s disease.
- Behcet’s disease can lead to blindness and early mortality due to vascular complications.
Reviewer
Dr Grainne Murphy
Consultant Rheumatologist
Cork University Hospital
Editor
Dr Chris Jefferies
References
- Tidy C, Cox J. Behcet’s Disease. Reviewed in 2016. Available from: [LINK]
- Adil A, Goyal A, Bansal P, Quint P. Behcet Disease. StatPearls. Updated in 2021. Available from: [LINK]
- Altiner A, Mandal R. Oral ulcer in patient with Behcet’s disease. Licence: [CC BY-SA 3.0]
- EyeMD. Anterior uveitis and hypopyon in a patient with Behcet’s disease. Licence: [CC BY-SA 2.5]
- Shaoon R, Daveluy S. Pathergy Test. StatPearls. Updated in 2021. Available from: [LINK]
- American Behcet’s Disease Association. Diagnosis of Behcet’s Disease. Published in 2018. Available from: [LINK]
- Haterni G, Christensen R, Bang D, Bodaghi B, Celik AF, Fortune F, et al. 2018 Update of the EULAR recommendations for the management of Behcet’s syndrome. Annals of the Rheumatic Diseases 2018;77:808-818. Available from: [LINK]
- National Health Service. Treatment: Behcet’s disease. Reviewed in 2019. Available from: [LINK]
