
- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Chronic myeloid leukaemia (CML) is a cancer of white blood cells that is characterised by the uncontrolled growth of myeloid cells in the bone marrow.
This uncontrolled growth leads to myeloid cells crowding out the bone marrow and interfering with the production of normal blood cells such as platelets, erythrocytes and neutrophils.
In addition to crowding out the bone marrow, the abnormal myeloid cells spill into the peripheral blood. This is reflected in the full blood count which demonstrates abnormally raised levels of mature granulocytes (neutrophils, basophils and eosinophils).
Aetiology
Pathophysiology
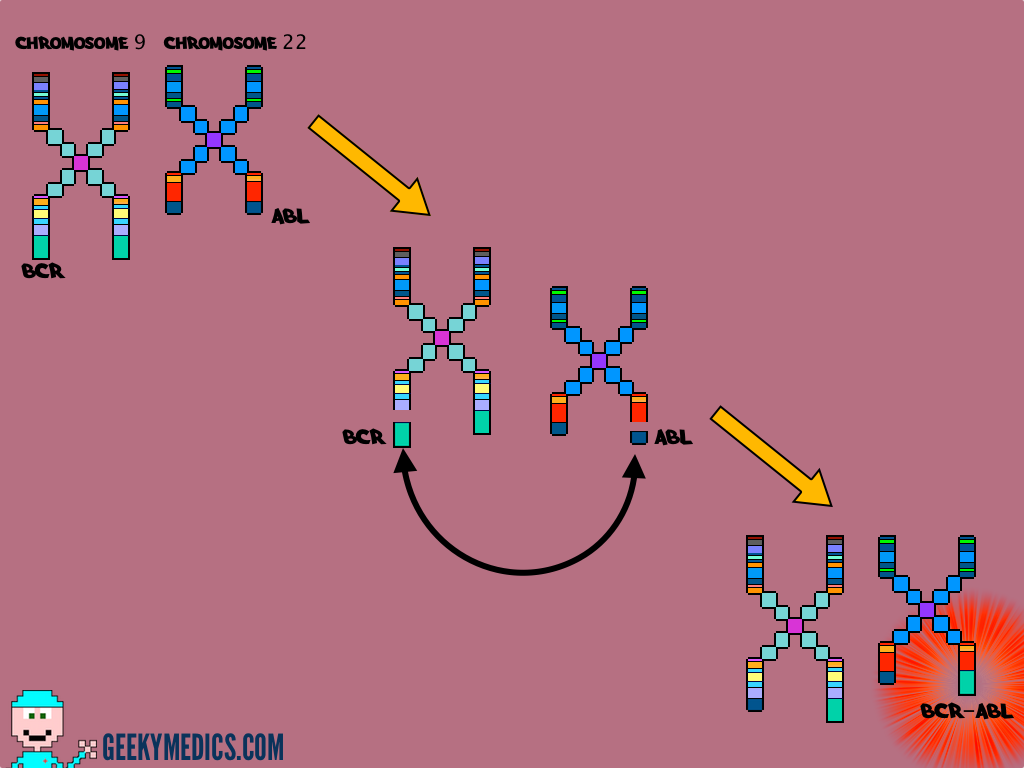
CML was the first malignancy to be linked directly to a clear chromosomal abnormality.
This abnormality involves a translocation between parts of chromosome 9 and 22, which leads to the formation of the so-called Philadelphia chromosome.
As a result, a gene contained on chromosome 22 called BCR fuses with a gene on chromosome 9 known as ABL. This creates a hybrid gene known as BCR-ABL.
BCR-ABL can add phosphates to tyrosine residues (a tyrosine kinase) and this allows the gene to activate a cascade of proteins that control the cell cycle, speeding up cell division.
In addition, the BCR-ABL protein inhibits DNA repair, causing genomic instability and making the cell more susceptible to developing further genetic abnormalities.
These two mechanisms (speeding cell division and inhibiting DNA repair) lead to the development of chronic myeloid leukaemia.
Clinical features
History
Patients are often asymptomatic when diagnosed with CML, presenting incidentally with an elevated white blood cell count on a full blood count.
Typical symptoms (if present) of CML include:
- Upper abdominal pain: due to hepatosplenomegaly
- Poor appetite: an enlarged spleen may compress the stomach
- Low-grade fever/night sweats (“B symptoms”)
- Gout: increased cell turnover → excess purines → broken down to uric acid
- Increased susceptibility to infections: decreased functional immune cells
- Shortness of breath/fatigue: anaemia
- Easy bruising/petechiae/bleeding: low platelets
- Neurological deficits/visual disturbance: hyperviscosity due to leucocytosis
Typical clinical findings include:
- Pallor
- Hepatosplenomegaly
- Bruising/petechiae
Investigations
Laboratory investigations
Relevant laboratory investigations include:
- Full blood count: leucocytosis, increased numbers of eosinophils and basophils, increased granulocytes, anaemia (normocytic/normochromic)
- U&E: usually normal, useful for a baseline prior to treatment
- Lactate dehydrogenase: may be raised
- Urate: may be raised due to high cell turnover
- Peripheral blood film: all stages of granulocyte maturation noted. Appearances are similar to that of a bone marrow aspirate.
Bone marrow aspirate
Bone marrow aspiration is necessary to stage disease (% of blasts determines chronic vs accelerated vs blast crisis) and enables cytogenetic sampling to confirm the diagnosis (presence of Philadelphia chromosome).
The gold standard of diagnosis is the detection of the Philadelphia chromosome, which is present in 95% of people with CML.
Methods for identifying the Philadelphia chromosome include FISH (fluorescent in-situ hybridisation) and PCR for the BCR-ABL gene.
Phases of CML
The World Health Organisation (WHO) has developed criteria to define the various stages of CML.¹
Chronic phase
Approximately 85% of patients with CML are in the chronic phase at the time of diagnosis.²
During this phase, patients are usually asymptomatic or have only mild symptoms of fatigue or abdominal fullness.
The duration of the chronic phase is variable and without treatment, the disease will usually progress to the accelerated phase due to the accumulation of genetic mutations in addition to the BCR-ABL gene.²
Accelerated phase
A patient is said to be in the accelerated phase when any of the following are present:
- 10 – 19% myeloblasts in the blood or bone marrow
- >20% basophils in the blood or bone marrow
- Platelet count <100,000, unrelated to therapy
- Platelet count >1,000,000, unresponsive to therapy
- Cytogenetic evolution with new abnormalities in addition to the Philadelphia chromosome
- Increasing splenomegaly or white blood cell count, unresponsive to therapy
This phase is significant as it is a sign that the disease is progressing and that a blast crisis is imminent.²
Blast crisis
Blast crisis is the final phase in the evolution of CML and behaves like acute leukaemia, with rapid progression and short survival.
Blast crisis is diagnosed if any of the following are present in a patient with CML:
- >20% myeloblasts or lymphoblasts in the blood or bone marrow
- Large clusters of blasts in the bone marrow on biopsy
- Development of a chloroma (a solid focus of leukaemia outside the bone marrow)
Management
Tyrosine kinase inhibitors
Tyrosine kinase inhibitors target BCR-ABL, blocking the ability of the gene to phosphorylate a tyrosine. This inhibits the proliferation of malignant cells.
Imatinib is a first-generation tyrosine kinase inhibitor and is usually the first-line treatment option. Alternatives include second-generation drugs including nilotinib and dasatinib.
Bone marrow transplantation
Bone marrow transplantation is a treatment option for those who do not respond well to drug treatments.
This tends only to be used in younger, otherwise healthy patients.
It does offer the possibility of a cure, however, there are serious risks (including death), therefore the treatment is not suitable for many patients.
References
- Vardiman J, Harris N, Brunning R (2002). The World Health Organization (WHO) classification of the myeloid neoplasms. Blood 100 (7): 2292–302. Available from: [LINK]
- Classification, diagnosis and management of myeloproliferative disorders in the JAK2V617F era”. Hematology Am Soc Hematol Educ Program 2006: 240–5.
