

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Cytochrome P450 (CYP450) are a group of enzymes encoded by the P450 genes and responsible for the metabolism of most drugs seen in clinical practice.
90% of drugs are metabolised by CYP3A5, CYP3A4, CYP2D6, CYP2C19, CYP2C9 and CYP1A2. CYP3A4 and CYP2D6 are the most significant enzymes.1
Polymorphism
Polymorphism is the genetic mutations that give rise to enzymes with different abilities to metabolise drugs. These genetic variabilities are responsible for the inter-individual variability in therapeutic response and toxicity to all major classes of drugs given at the standard dose.
The expression of CYP450 enzymes varies between populations and will greatly influence drug metabolism and response.
For example, CYP2D6 polymorphisms are expressed in four different phenotypes:
- Poor metabolisers (PM)
- Intermediate metabolisers (IM)
- Extensive metabolisers (EM)
- Ultrarapid metabolisers (UM)
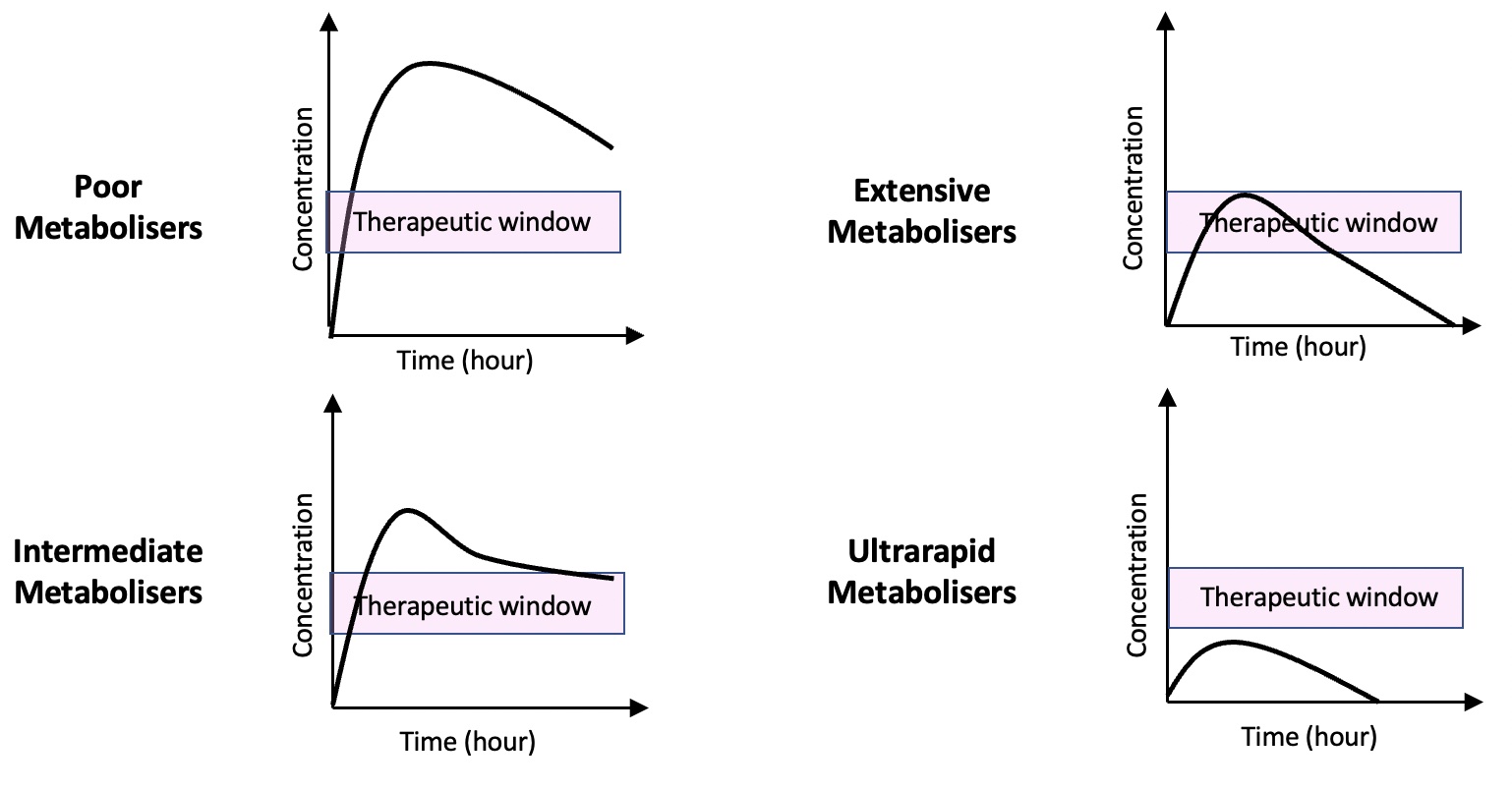
Poor metabolisers are characterised by the inability to metabolise drugs via the CYP2D6 metabolic pathway, resulting in an increased risk of adverse effects and toxicity. PM phenotype affects up to 10% of Caucasians and 30% of the Chinese population.2,3
At the other extreme, ultrarapid metabolisers metabolise the drug rapidly, resulting in a lack of therapeutic response in these individuals.
However, the reverse applies to prodrugs (drugs that are converted to their active forms in the body). Poor metabolisers fail to convert the prodrug into its active form leading to a lack of therapeutic response. In contrast, ultrarapid metabolisers rapidly convert the prodrug to its active form, causing potential toxicity.
Ultrarapid metaboliser phenotypes are most prevalent in the North African, Ethiopian and Arab populations, affecting 16% – 28% of the populations. In the rest of the world, the prevalence of ultrarapid metaboliser phenotypes is estimated to be 1% in the Chinese, Japanese and Hispanic populations and 5.5% in Western Europe.3,4
Example: Propranolol (a drug metabolised by CYP2D6)
Propranolol is a beta-blocker and a substrate of CYP2D6. In poor metabolisers, the metabolism of propranolol is greatly reduced. As a result, the higher plasma concentration of propranolol increases the risk of side effects and in some cases may lead to toxicity.
Signs and symptoms of beta-blocker overdose include light-headedness, dizziness, syncope, bradycardia and hypotension.
Example: Codeine (a prodrug metabolised by CYP2D6)
Codeine is a weak opioid and a substrate of CYP2D6. In ultrarapid metabolisers, codeine is metabolised more rapidly to its active compound, morphine as compared to individuals who are extensive metabolisers. Therefore, ultrarapid metabolisers may experience symptoms of opioid overdose (e.g. extreme drowsiness, respiratory depression and confusion) despite taking the standard dose.
Intermediate metabolisers have a reduced metabolism capacity compared to extensive metabolisers (who are classified as “normal”), therefore are more susceptible to adverse effects.
For example, nortriptyline is a common tricyclic antidepressant and a substrate of CYP2D6. In intermediate metabolisers, the metabolism of nortriptyline is reduced as compared to extensive metabolisers. As a result, the higher plasma concentration of nortriptyline in intermediate metabolisers increases the risk of potential side effects. A dose reduction should be considered in these patients.
CYP450 enzyme substrates, inducers and inhibitors
Substrates
Enzyme substrates are drugs or other substances that bind to and are metabolised by the CYP450 enzymes
Examples of CYP450 substrates include:
- Statins
- Theophylline
- Phenytoin
- Warfarin
- Selective serotonin reuptake inhibitors (SSRI): sertraline, citalopram, fluoxetine
- Amitriptyline
- Codeine
- Caffeine
Inducers
Inducers increase the expression level of CYP450 enzymes resulting in increased metabolism of drugs and subsequently reducing the therapeutic concentration.
Therefore, potential changes in drug concentration may cause treatment failure. The effects usually develop over several days and may be slow to resolve depending on the half-life of the inducer.
Examples of CYP450 inducers include:
- Anticonvulsants: phenytoin, carbamazepine, phenobarbitone
- Steroids: dexamethasone, prednisolone, glucocorticoids
- Antibiotics: rifampicin, griseofulvin
- Others: nicotine, alcohol, cigarette smoke, St John’s Wort
Inhibitors
Inhibitors prevent the CYP450 enzymes from working or reduce the rate of an enzyme-catalysed reaction. Consequently, this decreases drug metabolism in the body and increases the potential for toxicity. The effect often occurs quickly and is dose related.
Examples of CYP450 inhibitors include::
- Azoles: ketoconazole, fluconazole
- Antibiotics: sulfonamides, metronidazole, ciprofloxacin, chloramphenicol, macrolides, isoniazid
- Cimetidine
- Omeprazole
- Sodium valproate
- Grapefruit
Clinical relevance: Prescribing oral contraceptives for patients taking St John’s wort
St John’s wort is a CYP450 3A4 and 3A5 enzymes inducer. It increases the metabolism and clearance of oral contraceptive pills such as levonorgestrel, norethisterone, ethinylestradiol and desogestrel from the body. As a result, patients may experience breakthrough bleeding and potential contraceptive failure.
St John’s wort should not be taken concurrently with oral contraceptive pills or patients should use alternative methods such as barrier methods, depots and intrauterine devices (IUD).
For patients who require emergency contraception, a copper IUD is preferred over levonorgestrel. However, in cases where a contraindication arises for a copper IUD, 3 mg of levonorgestrel should be given as a single dose during and within 28 days after stopping St John’s wort.5
Clinical relevance: Prescribing miconazole for patients taking warfarin
Miconazole (commonly prescribed for oral thrush) is a CYP450 2C9 enzyme inhibitor. It inhibits the metabolism and clearance of warfarin, subsequently causing a rapid and extensive increase in warfarin concentration in the body. As a result, the anticoagulant effect of warfarin is increased, measured by an increase in the international normalised ratio (INR).
Miconazole should not be prescribed concurrently with warfarin. If necessary, monitor INR and reduce a patient’s warfarin dose accordingly. Patients should be advised to seek immediate medical attention if they experience any signs of bleeding, which include unexplained bruising, nose bleeds, or blood in their urine.5
Key points
- CYP450 enzymes are responsible for the metabolism of 90% of the drugs seen in clinical practice with CYP3A4 and CYP2D6 being the most significant enzymes
- Polymorphism of CYP450 enzymes has a huge impact on the inter-individual and interethnic variabilities in drug response and toxicity for a standard dose
- The clinical effects of CYP450 enzyme substrates, inducers and inhibitors should be kept in mind when prescribing as they can greatly influence prescribing therapy
Reviewer
Izaan Abd Rahman
Clinical Pharmacist
Editor
Dr Chris Jefferies
References
- Lynch T and Price A. The Effect of Cytochrome P450 Metabolism on Drug Response, Interactions, and Adverse Effects. Published in August 2007. Available from: [LINK]
- Zanger UM, Raimundo S and Eichelbaum M. Cytochrome P450 2D6: Overview and Update on Pharmacology, Genetics, Biochemistry. Published in November 2003. Available from: [LINK]
- Wong C, Lau E, Palozzi L and Campbell F. Pain management in children: Part 2 – A transition from codeine to morphine for moderate to severe pain in children. Published in November 2012. Available from: [LINK]
- Ingelman-Sundberg M. Genetic Polymorphisms of Cytochrome P450 2D6 (CYP2D6): Clinical Consequences, Evolutionary Aspects and Functional Diversity. Published in October 2004. Available from: [LINK]
- Royal Pharmaceutical Society. Stockley’s Drug Interactions via Medicines Complete. Available from: [LINK]
