

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Glomerulonephropathies are a set of diseases which cause inflammation in the glomeruli. These can be classified in a variety of different ways. This article will cover the causes of ‘nephrotic‘ and ‘nephritic‘ syndrome and the management of a patient with suspected glomerular disease.
Classification
Diseases of the glomerulus can be broadly divided into nephrotic syndrome and nephritic syndrome. A syndrome is a group of symptoms which consistently occur together.
In the case of nephrotic and nephritic syndromes, these are descriptive terms for a group of symptoms caused by different diseases. For example, a patient is not diagnosed with nephrotic syndrome. Instead, they are diagnosed with nephrotic syndrome caused by minimal change disease or membranous glomerulonephropathy.
Nephrotic syndrome is an important and well-defined clinical entity, and useful clinically to nephrologists treating patients.
The term ‘nephritic’ refers to glomerulonephritis (i.e. inflammation in the glomerulus), characterised by blood and protein present in the urine dip and often reduced kidney function. Nephritic syndrome is a less commonly used term clinically and refers to severe and usually acute presentations of glomerulonephritis causing hypertension and reduced urine output.
However, it is still helpful to think about nephritic syndrome to split the different types of glomerulopathy.
While some diseases generally cause either nephrotic syndrome or nephritic syndrome, many can overlap and have features of both. Some conditions may also present with mild proteinuria +/- haematuria that would not meet the nephrotic or nephritic syndrome definition.
Table 1. Nephrotic vs nephritic syndrome
| Nephrotic syndrome | Nephritic syndrome |
|
|
Diseases of the glomerulus can also be classified as primary or secondary.
Primary diseases affect the kidney directly, such as idiopathic (primary) membranous nephropathy. In contrast, secondary causes are when the kidney disease is driven by another disease or process (e.g. cancers, drugs, infections).
Pathology
The terminology used when describing the disease which affects the glomerulus can also be confusing.
They are used when describing the histological pattern of disease on kidney biopsy and are generally not specific diagnoses. The only real exception to this is focal segmental glomerulosclerosis (FSGS), a disease entity causing nephrotic syndrome defined by the presence of focal and segmental glomerulosclerosis on histology.
Terms used to describe glomerular disease:
- Focal: affecting some glomeruli
- Diffuse: affecting all glomeruli
- Segmental: affecting part of the glomerulus only
- Global: affecting the entire glomerulus
- Proliferative: increase in the number of cells in the glomerulus
- Crescent: a proliferation of cells within the bowman’s capsule that squashes the glomerular capillaries, giving a crescent appearance
Nephrotic syndrome
Nephrotic syndrome is typically caused by diseases which damage the filtration barrier at the glomerular basement membrane and is defined by the triad of:
- Proteinuria (>3.5gram protein/24hrs)
- Hypoalbuminemia
- Oedema
Clinical features1
Proteinuria
Nephrotic range proteinuria is defined as a loss of 3.5g of protein per 24 hours. Patients may describe passing frothy urine, or excess protein is detected on urine sampling. This equates to a urine protein:creatinine ratio (PCR) of approximately 300mg/mmol.
Proteinuria is caused by increased basement membrane permeability, causing increased urinary loss of protein (including albumin).
Hypoalbuminemia
Loss of albumin in the urine. Usually, the liver would be able to compensate by increasing albumin production. However, for unknown reasons, the liver cannot compensate in patients with nephrotic syndrome.
Oedema2
Classically, oedema is thought to be related to hypoalbuminemia, causing reduced plasma oncotic pressure and, therefore, fluid movement to the extracellular compartment. More recently, it is believed to be related to excess salt and water retention in the kidney triggered by nephrotic syndrome. Patients present with gross peripheral oedema.
Other clinical features
Other clinical features of nephrotic syndrome may include:
- Hyperlipidaemia: increased hepatic production of triglycerides. This is poorly understood but thought to be due to hypoalbuminemia and changes to albumin-bound lipids. Hyperlipidaemia leads to an increased cardiovascular risk profile and can cause xanthoma.
- Thrombosis: caused by loss of antithrombotic factors in the urine and increased production of prothrombotic factors in the liver. Patients are at increased risk of VTE and often require anticoagulation.3
- Immunodeficiency: due to loss of immunoglobulins such as IgG and other immune proteins in the urine.
- Renal impairment and hypertension: intrinsic damage to the kidney from the underlying disease process. The presence/degree of renal impairment depends on the disease.
Predominantly nephrotic disease
Table 2. Conditions which predominantly cause nephrotic syndrome.4,5
| Disease | Overview | Aetiology | Histology | Treatment |
|
Minimal change nephropathy |
The most common cause of nephrotic syndrome in children. Typically presents around 2 years old with facial swelling. Abrupt onset
|
Primary or secondary to things such as NSAIDs/lymphoma/allergy. Several genetic causes are recognised and are often refractory to treatment. |
Light microscopy normal. Electron microscopy shows podocyte foot process effacement. |
Usually very steroid responsive but can relapse. May require further immunosuppression. |
|
Focal segmental glomerulosclerosis |
Can progress to chronic renal failure. |
Primary or secondary to HIV, drugs (e.g. pamidronate), urinary reflux, sickle cell disease. Genetic factors such as APOL-L1 mutations are associated with an increased risk of FSGS and progression to ESKD |
Focal collapse and sclerosis. Patchy involvement and may be missed on biopsy. Different histopathological subtypes have varying prognoses. |
Not always steroid responsive. May require immunosuppression, renal replacement therapy and transplantation. Can recur in a transplant kidney. |
|
Membranous glomerulo-nephropathy |
The commonest cause of nephrotic syndrome in adults. |
Primary or secondary. 70% of primary cases are associated with anti-PLA2R antibodies. Other causes include malignancy, drugs, infections (e.g. viral hepatitis) or SLE. |
Subepithelial deposition of immune complexes with basement membrane thickening – produces a spiked appearance on silver stain. |
The prognosis depends on the cause. May remit spontaneously, but otherwise may need immunosuppression. |
|
Amyloidosis |
Amyloidosis involves extracellular protein deposits. This can deposit in multiple organs including the kidney. |
Broadly split into AA Amyloid (associated with chronic inflammation) and AL amyloid (associated with haematological disorders). There are also a few rare genetic causes of amyloidosis. |
Amyloid stains with Congo red, with apple-green birefringence on polarised light microscopy. |
Depends on the underlying cause but renal amyloidosis generally carries a poor prognosis |
|
SLE (lupus) nephritis (specifically class V subtype) |
Can present multiple histological findings and features of both nephrotic and nephritic syndromes. | |||
Management
Treatment varies depending on the cause of nephrotic syndrome. However, treatment can be divided into general/supportive measures and immunosuppression.
General/supportive measures for nephrotic syndrome include:9
- Salt and fluid restriction, diuretics (typically furosemide in the first instance): to reduce oedema
- Commence ACE inhibitor/ARB: to reduce proteinuria
- Consider VTE prophylaxis: due to hypercoagulability
- Pneumococcal vaccine: due to immunosuppression
- Commence statin: to manage dyslipidaemia and reduce cardiovascular risk
Unless the patient is deemed high-risk, supportive measures may be tried to see if remission occurs without the need for immunosuppression.
Immunosuppression regimens vary depending on the cause of nephrotic syndrome:
- Minimal change disease and FSGS: usually initially treated with steroids (prednisolone): further immunosuppression is considered if failure to achieve remission or if there is a relapse
- Membranous nephropathy: treatment depends on the severity of the disease, but may include steroids and cyclophosphamide, or rituximab
Other agents that may be used (particular if treating resistant or relapsing diseases) include calcineurin inhibitors (e.g. tacrolimus), mycophenolate mofetil, azathioprine and rituximab.
Nephritic syndrome
Nephritic syndrome occurs due to inflammation and damage to endothelial cells of glomerular capillaries and can be triggered by several disease processes.
Clinical features6
Clinical features of nephritic syndrome include:
- Proteinuria: this is typically, but not always, in the sub-nephrotic range
- Haematuria: can be micro or macroscopic haematuria. It can cause red cell casts which can be examined under microscopy.
- Hypertension: this occurs due to salt and water retention, in part mediated by glomerular damage causing activation of the renin-angiotensin-aldosterone (RAAS) system. Typically will occur in more acute or severe cases. Hypertension can be so severe it can result in hypertensive encephalopathy.7
- Renal impairment & oliguria: oliguria occurs as a reduction in glomerular filtration, in severe or advanced cases.
- Pyuria: can be present and will be sterile (i.e. no organisms present). This indicates inflammation and can form white cell casts in the urine that can be examined under microscopy.
Chronicity of disease is important in nephritic syndrome. If symptoms occur and renal function declines over days or weeks, it can be classified as acute glomerulonephritis. If the symptoms occur over months to years with little reduction in kidney function, then it is chronic glomerulonephritis.
Rapidly progressive glomerulonephritis (RPGN)
Rapidly progressive glomerulonephritis (synonymous with crescentic GN) is a sub-classification of acute glomerulonephritis which is associated with crescents on renal biopsy.
The definition typically involves a 50% decline in kidney function over 3 months but can be much quicker (i.e. over a matter of days/weeks).
RPGN is not a single disease process but describes glomerulonephritis, which presents very acutely with a rapid decline in kidney function and the presence of crescents (inflammation and proliferation of cells in the Bowman’s space) compresses the glomerulus, giving the appearance of crescents) on histology.6
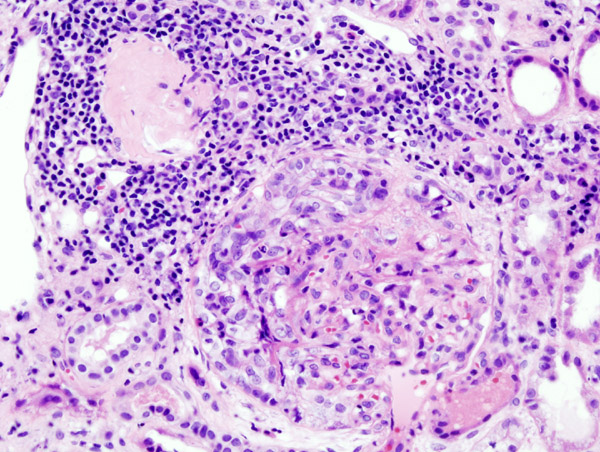
Pathology
The causes of acute glomerulonephritis can also be subclassified into three main pathologies: immune complex disease, vasculitis (often pauci-immune) and anti-GBM associated diseases.
Immune complex diseases
Immune complex diseases are caused by an activation of the complement system by immune complexes that deposit on the glomerular capillaries, leading to inflammation and damage. They are characterised by low C3 and/or C4 levels, depending on which arm of the complement system is activated.
Diseases include post-infectious glomerulonephritis (usually post-streptococcal infection) and membranoproliferative glomerulonephritis (MPGN). MPGN is a histological finding and can be caused by paraprotein-producing malignancies, autoimmune diseases and chronic viral infections (e.g. hepatitis C).
Pauci-immune vasculitis
Vasculitis can also cause nephritic syndrome. Vasculitis is a systemic disease involving other organ systems such as the lung or skin but can involve the kidneys, or be renal-specific. Complement levels will be normal.
Vasculitis can be split into small, medium or large vessel disease or be subclassified according to their immunofluorescence pattern on histology.
Diseases typically associated with renal vasculitis are granulomatosis with polyangiitis (GPA), eosinophilic granulomatosis with polyangiitis (eGPA) and microscopic polyangiitis (MPA).
The term ‘pauci-immune’ refers to the relative lack of immune deposits on immunofluorescence.
These are associated with ANCA antibodies which are often helpful in monitoring treatment response:
- GPA is associated with cANCA (anti-PR3)
- MPA is associated with pANCA (anti-MPO)
Anti-GBM disease
Anti-GBM disease can cause nephritic syndrome, typically an RPGN. The complement levels will be normal. This is caused by anti-GBM antibodies against the type IV collagen present in the kidneys and the lungs.
If the disease is associated with pulmonary and renal features it is called Goodpasture’s disease, however, anti-GBM can be kidney specific. Classically there is linear GBM staining on immunofluorescence. Lung involvement may present as massive haemoptysis.
Predominantly nephritic disease
Table 3. Conditions which predominantly cause nephritic syndrome10
| Immune complex diseases | |
|
Post-streptococcal GN |
Pathology: due to group A streptococcal infection (GAS). Clinical features: low C3/4. Positive ASOT. Presents 2-3 weeks after GAS infection. Nephritic symptoms. Treatment: treat GAS infection. |
|
IgA GN (previously Berger’s disease) |
Pathology: due to IgA deposition. Associated with Henoch-Schonlein purpura. Clinical features: typically haematuria hours to days after a recent respiratory tract infection. Can present with just haematuria or nephritic syndrome. Raised plasma IgA and IgA deposition with mesangial expansion on renal biopsy. Treatment: ACE inhibitors, consider immunosuppression in specific cases. |
|
Membranoproliferative glomerulonephritis (MPGN)8 |
Pathology: basement membrane and mesangial thickening & proliferation on light microscopy with “tram tracking” due to GBM expanding over the top of immune deposits. Clinical features: nephritic or nephrotic clinical picture. Associated with a number of conditions including haematological malignancies, autoimmune diseases, chronic viral infections, and genetic abnormalities in the complement system. Treatment: treat the underlying cause (if secondary), with immunosuppressive therapies. |
|
Others |
|
| Pauci-immune diseases | |
|
Granulomatosis with polyangiitis:
Eosinophilic granulomatosis with polyangiitis:
Microscopic polyangiitis:
|
|
| Anti-GBM associated diseases | |
|
Anti-GBM disease and Goodpasture’s syndrome |
Pathology: caused by antibodies against the type IV collagen which is located in the GBM and alveolar capillaries. Clinical features: either reno-specific or affecting the lungs. If the lungs are affected, it is Goodpasture’s syndrome. RPGN picture usually. Lung involvement may manifest as massive haemoptysis. Treatment: steroids, cyclophosphamide, plasma exchange |
Management
Like nephrotic syndrome, treatment varies depending on the underlying cause and severity.
For example, many patients with IgA nephropathy have a slowly progressive course where the only treatment is supportive (e.g. ACE inhibitors for control of blood pressure and proteinuria). Conversely, anti-GBM disease typically presents as an RPGN requiring immediate and intensive immunosuppression and plasma exchange.
For more information, see the Geeky Medics guide to glomerulonephritis.
If severe renal impairment then dialysis may be required. Transplantation may be considered later as long as the disease process is controlled. For more information, see the Geeky Medics guide to chronic kidney disease.
Key points
- Glomerulonephropathies are a set of diseases which affect the glomeruli.
- Nephrotic syndrome typically presents with proteinuria, hypoalbuminemia and oedema.
- Nephritic syndrome typically presents with proteinuria, haematuria, hypertension and oliguria (in severe cases).
- While some glomerular diseases generally cause either nephrotic syndrome or nephritic syndrome, there are many which can overlap and have clinical features of both.
- The management of glomerular disease depends on the underlying disease process but may involve immunosuppression.
Reviewer
Dr Mohammed Al-Talib
Renal Registrar
Editor
Dr Chris Jefferies
References
- Macé C, Chugh SS. Nephrotic syndrome: components, connections, and angiopoietin-like 4-related therapeutics. J Am Soc Nephrol. 2014 Nov;25(11):2393-8.
- Rondon-Berrios H. New insights into the pathophysiology of oedema in nephrotic syndrome. Nefrología (English Edition). 2011 Mar 1;31(2):148-54.
- Al-Azzawi HF, Obi OC, Safi J, Song M. Nephrotic syndrome-induced thromboembolism in adults. Int J Crit Illn Inj Sci. 2016 Apr-Jun;6(2):85-8.
- Nephcure. FSGS. 2022. Available from: [LINK]
- Nephcure. Membranous nephropathy. 2022. Available from: [LINK]
- Saint, Sanjay and others (eds), ‘Nephritic Syndrome’, in Sanjay Saint, and Vineet Chopra (eds), The Saint-Chopra Guide to Inpatient Medicine, 4 edn (New York, 2018; online edn, Oxford Academic, 1 Nov. 2018)
- Ihm CG. Hypertension in Chronic Glomerulonephritis. Electrolyte Blood Press. 2015 Dec;13(2):41-5.
- National Kidney Foundation. Membranoproliferative Glomerulonephritis. Available from: [LINK]
- Oxford Handbook of Clinical Specialities, 9th Ed. 2013.
-
Lionaki, S. , Skalioti, C. , Marinaki, S. , N. Boletis, J. . Pauci-Immune Vasculitides with Kidney Involvement. In: Mohammed, R. H. A. , editor. Vasculitis In Practice – An Update on Special Situations – Clinical and Therapeutic Considerations [Internet].
Image references
- Figure 1. KGH.Crescentic glomerulonephritis. License: [CC BY-SA]
