

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Polycythaemia vera is a myeloproliferative disorder characterised by the excess production of erythrocytes. In over 98% of cases, it is caused by a genetic mutation in the JAK2 gene.1
Polycythaemia vera is a rare condition with an incidence of 2 per 100,000 and a prevalence of 50 per 100,000. It tends to present in patients aged 60 – 70 years old.2
Aetiology
Myeloproliferative disorders result in the excess production of myeloid cells: erythrocytes (red blood cells), platelets or granulocytes (neutrophils, eosinophils and basophils). When excess red blood cells are produced resulting in a raised haemoglobin concentration and haematocrit this is known as polycythaemia.
Polycythaemia vera is a primary polycythaemia. In almost all cases, the driver of the excess erythrocyte production is a mutation in the JAK2 (Janus Kinase 2) gene.1
This contrasts with secondary polycythaemia where a high number of erythrocytes are produced in a physiological response to chronic hypoxia (i.e. secondary to smoking or chronic lung disease), local renal hypoxia (e.g renal artery stenosis) or excess erythropoietin (EPO) production (i.e. secondary to EPO secreting tumours).2
Risk factors
Risk factors for polycythaemia vera include:
- Advancing age: median age at diagnosis is 60-70 years
- History of Budd-Chiari syndrome: a proportion of people with what had previously been identified as idiopathic Budd-Chiari syndrome (or other splanchnic vein thromboses), will in fact have a JAK2 mutation even if they had initially normal blood counts
Clinical features
History
Typical symptoms of polycythaemia vera may include:2,3
- Headaches: usually associated with dizziness and sweating
- Myalgia and weakness
- Fatigue
- Tinnitus
- Pruritis: particularly after a hot shower or bath
- Erythromelalgia: burning pain, warmth and redness in the hands and feet
- Blurred vision: temporary loss of vision due to hyper-viscosity
- Dyspepsia: peptic ulceration
- Gout: due to increased cell turnover
A third of patients first present due to thrombosis including stroke, myocardial infarction, deep vein thrombosis, pulmonary embolism, Budd-Chiari syndrome. In patients with polycythaemia vera, 75% of thromboses are arterial, and 25% are venous.
Clinical examination
Typical clinical findings of polycythaemia vera include2,3:
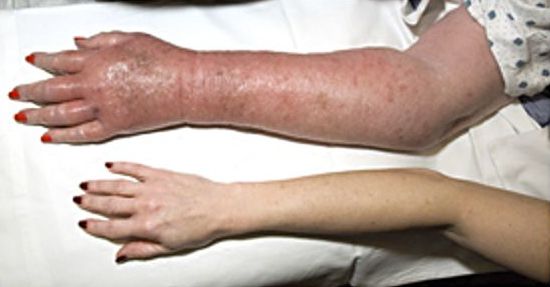
- A ‘ruddy’ (reddish) complexion
- Splenomegaly: present in one-third of patients at the time of diagnosis
- Abdominal masses: benign and malignant uterine, renal and hepatic tumours which can secrete EPO may be palpable
- Hypertension
Differential diagnoses
In the context of a raised haemoglobin, differential diagnoses to consider include:5
- Hypoxia driven: including COPD, smoking, carbon monoxide poisoning, sleep apnoea, left to right cardiac shunts (Eisenmenger’s syndrome) and high altitude
- Local renal hypoxia: for example, secondary to renal artery stenosis
- Pathological erythropoietin production: secondary to EPO secreting tumours; including a clear cell renal carcinoma, Wilms’ tumour, hepatocellular carcinoma, cerebellar haemangioblastoma, pheochromocytoma, uterine myoma, parathyroid carcinoma and meningioma
Investigations
Full blood count
Polycythaemia is defined as:2,5
- Haemoglobin (Hb) >185 g/L and/or haematocrit (Hct) > 0.52 in males
- Hb >165 g/L and/or Hct > 0.48 in females
- Red cell mass >25% above predicted
If the patient is dehydrated, apparent polycythaemia may be present in which the Hb/Hct is raised because of a reduced plasma volume. These patients will have a normal red cell mass.5
One-off values of Hct >0.6 in males and >0.52 in females require additional investigation.
In addition, neutrophilia and thrombocytosis are commonly seen in patients with polycythaemia vera.
Other laboratory investigations
Other relevant laboratory investigations include:
- Blood film: to assess for features of leukaemia which require bone marrow assessment
- U&E/LFTs: to assess for renal/hepatic causes (including tumours) of secondary polycythaemia or complications (including Budd-Chiari syndrome) of polycythaemia vera
- Serum ferritin: normal or low in polycythaemia vera due to increased demand for iron
- Arterial blood gas: may identify hypoxia or raised carboxyhaemoglobin levels secondary to smoking or carbon monoxide poisoning5
- Serum erythropoietin: suppressed levels suggest polycythaemia vera whilst raised levels in the context of polycythaemia suggest a secondary cause; inappropriate EPO production and the possibility of an EPO-secreting tumour.
- JAK 2 V617F mutational analysis: positive in 95% of patients with polycythaemia vera. Around 5% of cases involve other JAK2 mutations.
Bone marrow biopsy
A bone marrow biopsy may be helpful in distinguishing polycythaemia vera from secondary polycythaemia. In polycythaemia, a biopsy may show hypercellularity, increased erythropoiesis, granulopoiesis and megakaryopoiesis, and variable megakaryocyte size.
Imaging
Relevant imaging investigations include:
- Abdominal ultrasound: to assess for splenomegaly and exclude secondary causes of polycythaemia including renal and hepatic pathology.
- Further imaging: CT head/neck/chest/abdomen/pelvis looking for rarer tumours which may secrete EPO. This is not always required if a cause for the polycythaemia is found with the above initial tests.
Diagnosis
Table 1. A summary of the diagnostic criteria as per the British Society of Haematology.5
| JAK2‐positive polycythaemia vera (requires both criteria) | |
|
A1 |
High haematocrit (>0·52 in men, >0.48 in women) OR raised red cell mass (>25% above predicted) |
|
A2 |
Mutation in JAK 2 |
| JAK2‐negative polycythaemia vera (requires A1‐A4 plus another A or two B criteria) | |
|
A1 |
Raised red cell mass (>25% above predicted) OR haematocrit ≥0.60 in men, ≥0.56 in women |
|
A2 |
Absence of mutation in JAK 2 |
|
A3 |
No cause of secondary erythrocytosis |
|
A4 |
Bone marrow histology consistent with polycythaemia vera |
|
A5 |
Palpable splenomegaly |
|
A6 |
Presence of an acquired genetic abnormality (excluding BCR‐ABL1 ) in the haematopoietic cells |
|
B1 |
Thrombocytosis (platelet count >450 × 109 /L) |
|
B2 |
Neutrophil leucocytosis (neutrophil count >10 × 109 /L in non‐smokers, ≥12·5 × 109 /L in smokers) |
|
B3 |
Radiological evidence of splenomegaly |
Management
Patients with polycythaemia vera are at increased risk of arterial and venous thrombosis and therefore initial management involves cardiovascular risk factor optimisation (i.e. hyperlipidaemia, diabetes, hypertension and smoking status is assessed and managed as appropriate).
Further treatment options aim to significantly reduce thrombosis risk whilst also reducing the risk of haemorrhage (which can be seen despite high platelet counts) and symptom burden.2,5
Phlebotomy
Intermittent long-term phlebotomy is performed to maintain Hct <0.45. Approximately 200-500mls of blood is venesected at intervals suitable to the patient.5
Aspirin
Low dose aspirin reduces the risk of thrombotic events and death in patients with polycythaemia vera. It does not increase the risk of haemorrhage.
Cytoreductive therapy
Cytoreductive therapy is indicated in those with:
- Progressive splenomegaly
- Progressive leucocytosis (WCC>15)
- Progressive thrombocytosis (platelets >1500)
- Poor tolerance to venesection (increased frequency of venesection required to maintain Hct<0.45)
- Age of 60 years and above
- History of thrombosis
Hydroxycarbamide is the first line cytoreductive agent. Interferon-alfa is a second line option or alternative if hydroxycarbamide is contraindicated, not tolerated or ineffective.
Interferon Alfa is particularly useful in women of childbearing age as it is not teratogenic which is a concern with hydroxycarbamide.2
Complications
Complications of polycythaemia vera include:3
- Ischaemic stroke
- Myocardial infarction
- Pulmonary embolism
- Progression to myelofibrosis or acute myeloid leukaemia
- Gastrointestinal haemorrhage
- Budd-Chiari syndrome
Prognosis
The median survival is approximately 14 years. Mortality is commonly related to thromboembolic events (ischaemic stroke and myocardial infarction).3
Progression to myelofibrosis at 15 years is up to 15%. Leukaemic transformation is rarer with 10% transformation at 20 years.2
Key points
- Polycythaemia vera is a myeloproliferative disorder characterised by the excess production of erythrocytes.
- Polycythaemia vera is the cause of primary erythrocytosis.
- In almost all cases, polycythaemia vera is caused by a genetic mutation in the JAK2 gene.
- It is usually diagnosed by the presence of a raised haematocrit OR red cell mass plus the presence of the JAK2 mutation.
- Patients with polycythaemia vera are managed with regular venesection, aspirin and in many cases cytoreductive agents including hydroxycarbamide, or interferon alfa.
- Management also involves the optimisation of cardiovascular risk factors.
- The main complications of polycythaemia vera are thrombotic events, haemorrhagic events and progression to acute myeloid leukaemia or myelofibrosis.
Reviewer
Dr Alex Langridge
Haematology Registrar
Editor
Dr Chris Jefferies
References
- Tefferi A et al. Polycythemia vera treatment algorithm 2018. Published in 2018. Available from: [LINK]
- NICE CKS. Polycythaemia/erythrocytosis. Revised in 2020. Available from: [LINK]
- Patient UK. Polycythaemia Rubra Vera. Revised in 2016. Available from: [LINK]
- Herbert L et al. Erythromelalgia. Licence: [CC-BY]
- McMullin MF et al. A guideline for the diagnosis and management of polycythaemia vera. A British Society for Haematology Guideline. Published in 2019. Available from: [LINK]
