

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Pyruvate kinase deficiency is an inherited metabolic condition affecting the glycolytic pathway.
The glycolytic pathway is present in all cells but is particularly vital in red blood cells where it is the main route to ATP production in the absence of mitochondria. When this pathway is interrupted, red blood cells distort and are removed from the circulation, leading to haemolytic anaemia.
Prevalence of pyruvate kinase deficiency, estimated from gene frequency, is thought to be 1 in 20,000 people, although the clinically observed frequency is far lower than this.1
Pyruvate kinase deficiency affects men and women equally and has been noted to be more prevalent in Europe, although this may be due to underdiagnosis and misdiagnosis of this rare disease.2
Aetiology
Most cases of pyruvate kinase deficiency are inherited in an autosomal recessive manner.
Autosomal dominant inheritance has occasionally been seen and compound heterozygotes (patients heterozygous for two different PKLR mutations) have been reported.
Pyruvate kinase deficiency can be acquired, either secondary to other haematological conditions, such as acute leukaemia or refractory sideroblastic anaemia, or as an adverse effect to chemotherapeutic agents.
Pathophysiology
Pyruvate kinase deficiency arises due to a variety of mutations in the protein kinase isoenzymes (PKLR) gene.
PKLR is expressed in red blood cells (RBC) and catalyses the final reaction in the glycolysis pathway, converting phosphoenolpyruvate into pyruvate and phosphorylating ADP to make ATP (Figure 1).
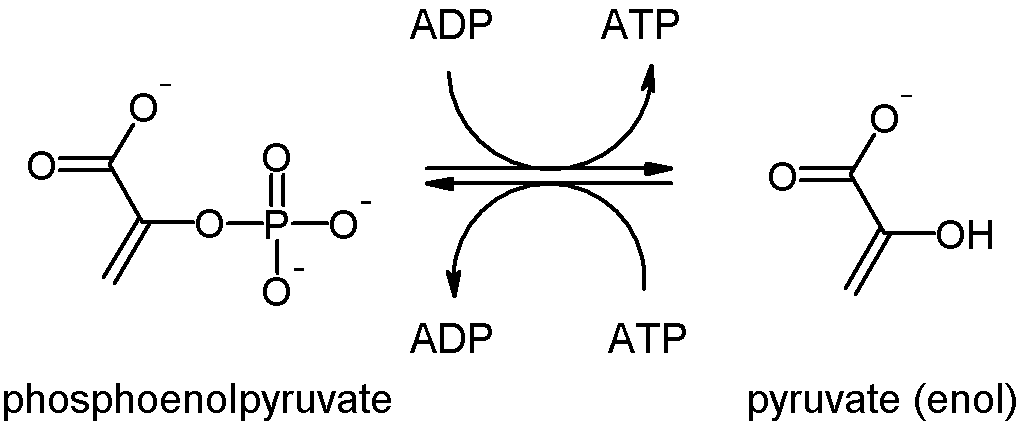
This mutation causes loss of enzymatic function, so red blood cells can no longer produce ATP from glycolysis. Without ATP, the membrane transporter sodium-potassium adenosine triphosphatase slows down, reducing ion influx into the cell.
The cell becomes hypotonic, so water leaves the cell down the osmotic gradient, causing the cell structure to shrink and distort. Damaged red blood cells are removed from the circulation by the reticuloendothelial system, which removes cells quicker than they can be produced, resulting in haemolytic anaemia.
Premature red blood cells, called reticulocytes, are released from the strained bone marrow tissue in response to increased RBC destruction.
Reticulocytes can survive in circulation as they have mitochondria, so can produce ATP by oxidative phosphorylation. They have a reduced oxygen-carrying capacity when compared to mature RBCs but can compensate for mild anaemia.
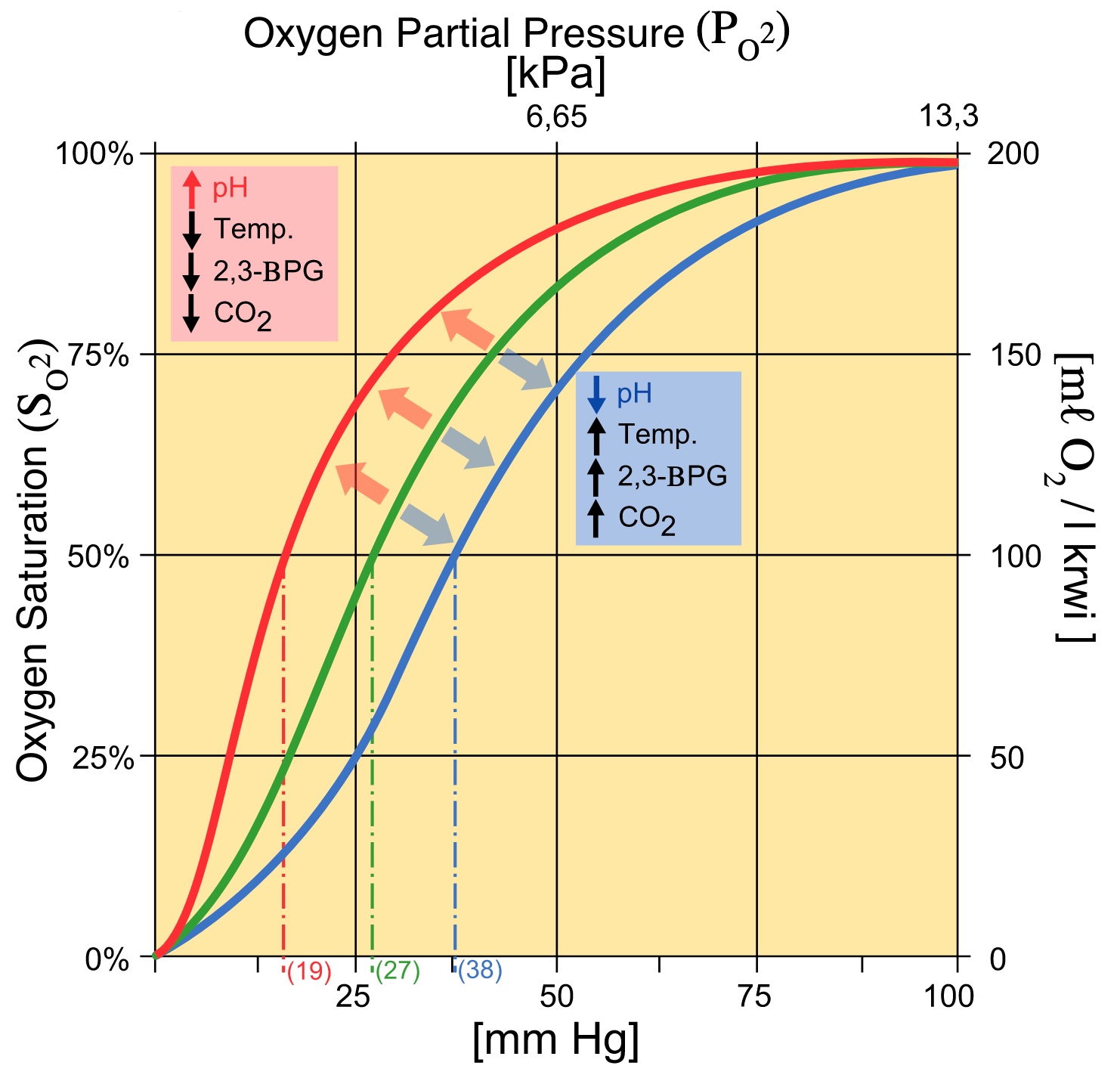
As well as a deficit in ATP and energy, this mutation causes a build-up of intermediates earlier in the glycolysis pathway. Accumulation of 2,3-BPG, causes a shift in the Hb-O2 dissociation curve to the right, increasing oxygen release at the tissues. This allows patients to further compensate for anaemia.
Clinical features
History
Symptoms from pyruvate kinase deficiency can be highly variable.
The most common presentation is symptomatic haemolytic anaemia which is often limited to childhood but can also occur during periods of stress, such as during pregnancy, surgery or infection.
The presentation of anaemia can range from very mild (compensated) anaemia to life-threatening haemolysis.
Typical symptoms of anaemia may include:
- Pallor
- Fatigue
- Dizziness
- Chest pain
- Dyspnoea
- Growth delay and failure to thrive (children)
Rarely, affected foetuses can develop hydrops fetalis resulting from the cardiac strain of severe foetal anaemia. In neonates, haemolysis can lead to high levels of bilirubin which can cause neurotoxicity leading to kernicterus.
Clinical examination
Clinical examination may reveal signs of haemolytic anaemia including:
- Jaundice: sclera often turns yellow first (icteric sclera), then mucous membranes and skin
- Splenomegaly and hepatomegaly: as the spleen and liver contain much of the reticuloendothelial system
- Tachycardia: early response to poor oxygen delivery to the body
- Dark urine or stool
- Chronic leg ulcers: poor perfusion to these areas causes delayed healing

Differential diagnoses
It is important to distinguish this condition from other causes of haemolytic anaemia including:
- Sickle cell anaemia
- Thalassaemia
- G6PD deficiency
- Hereditary spherocytosis and elliptocytosis
- Autoimmune disorders
- Infection: including Epstein-Barr virus, hepatitis or typhoid
- Medicines: including some antibiotics, quinidine, ibuprofen or mismatched blood transfusions
Investigations
Laboratory investigations
Relevant laboratory investigations include:
- Full blood count: normal Hb, low RBC, high MCV (known as normochromic macrocytic anaemia); can have a slight increase in WCC and platelets also
- Reticulocyte count: raised due to increased RBC production
- Blood film: signs of accelerated erythropoiesis, such as polychromasia and nucleated red blood cells. Prickle cells can also be seen particularly if patients have had a splenectomy.
- Unconjugated bilirubin and iron: elevated due to increased haemoglobin breakdown
It is also possible to test for proteins involved in the pathology, such as:
- Pyruvate kinase level
- 2,3-BPG level
- Glucose-6-phosphate level: to rule out G6PD deficiency
Molecular genetic testing for PKLR gene mutation is possible at specialist centres.
Management
Most cases of pyruvate kinase deficiency are mild and require little treatment.
However, severe disease can require hospitalisation to treat life-threatening anaemia.
Management options include:6
- RBC transfusion: may need to be frequent and regular to survive
- Splenectomy: slows breakdown of premature reticulocytes
- Allogenic haematopoietic stem cell transplant: can cure pyruvate kinase deficiency, although carries significant risk and a high mortality rate
Clinical trials are continuing to find new ways to treat pyruvate kinase deficiency, including small molecule activators and gene therapy.
The severity of the disease is judged based on symptoms rather than serum haemoglobin level, as the degree of anaemia compensation varies between patients.
Complications
Gallstones
High bilirubin levels can lead to gallstone production. The liver normally conjugates bilirubin with glucuronic acid, increasing the water-solubility of this compound, aiding its excretion from the body.
As bilirubin levels increase vastly, unconjugated bilirubin builds up, which binds with calcium ions to form insoluble precipitates. These precipitates can then crystallise together and form gallstones.
Gallstones may have no consequence but can lead to biliary colic, cholecystitis, cholangitis and acute pancreatitis.
Iron overload
Pyruvate kinase deficiency patients can experience iron overload, due to dyserythropoiesis or frequent blood transfusions.
Iron overload is asymptomatic in most patients, although serum ferritin levels should be monitored as chronic iron overload can result in organ damage:
- Liver: hepatomegaly, abdominal pain and eventual fibrosis
- Heart: arrhythmias and cardiac failure
- Endocrine organs: hypogonadism, decreased libido, hyperglycaemia and hyperpigmentation
Treatment of iron overload is typically venesection, although this is poorly tolerated in anaemic patients who may require the use of iron chelation therapies.
Other complications
Other less common complications of pyruvate kinase deficiency are osteoporosis, skin ulceration, pulmonary hypertension, aplastic crisis (secondary to parvovirus B19 infection) and extramedullary haematopoiesis.2
Transfusion of blood products, while a necessary treatment for many pyruvate kinase patients, can lead to complications, including transfusion reactions.
Key points
- Pyruvate kinase deficiency is an inherited metabolic condition affecting the glycolytic pathway.
- Most cases of pyruvate kinase deficiency are inherited in an autosomal recessive manner.
- Mutations in the PKLR gene causes disturbance of red cell function, leading to a distorted red cell structure.
- Abnormal red blood cells are removed from the circulation, causing the clinical features of haemolytic anaemia.
- Many cases do not require treatment, but transfusion, splenectomy and allogeneic stem cell transplants are sometimes used in the management of these patients.
- Pyruvate kinase deficiency patients can experience iron overload, due to dyserythropoiesis or frequent blood transfusions.
Reviewer
Dr Matthew Player
Haematology SpR
Editor
Dr Chris Jefferies
References
- Beutler E., Gelbart T.. Estimating the prevalence of pyruvate kinase deficiency from the gene frequency in the general white population. Published in 2000. Available from: [LINK]
- National Organization for Rare Disorders. Pyruvate Kinase Deficiency. Published in 2019. Available from: [LINK]
- Akane. Glycolysis PEP-Pyr. Licence: [CC-BY-SA].
- Komorniczak M., Herráez A. Adapted by Geeky Medics. Influence on Saturation Hemoglobin (HbA) curve. Haldane and Bohr Effect. Licence: [CC-BY-SA]
- Centers for Disease Control and Prevention (CDC). Yellowing of the skin and eyes. Licence: [Public domain]
- van Straaten S., Bierings M., Bianchi P., Akiyoshi K., Kanno H., Serra I.B., Chen J., Huang X., van Beers E., Ekwattanakit S., Güngör T., Kors W.A., Smiers F., Raymakers R., Yanez L., Sevilla J., van Solinge W., Segovia J.C., van Wijk R.. Worldwide study of hematopoietic allogeneic stem cell transplantation in pyruvate kinase deficiency. Published in 2018. Available from: [LINK]
