

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Retinitis pigmentosa (RP) encompasses a clinically and genetically diverse group of inherited retinal disorders that cause retinal degeneration. RP is the most common hereditary retinal dystrophy which causes progressive vision loss. In the UK, the prevalence of RP is approximately 1 in 4,000.1
Causes and risk factors
RP is caused by mutations in genes encoding proteins important for photoreceptor and retinal pigment epithelium (RPE) function and survival. Most cases result from mutations of the rhodopsin gene.
Genetic predisposition is the principal risk factor for RP. It may be sporadic or inherited in an autosomal dominant, autosomal recessive or X-linked pattern (Table 1).2 The prognosis is related to the mode of inheritance.
While RP is a disease usually limited to the eye, it can be associated with rare systemic syndromes (syndromic RP) with the most common being Usher syndrome (US) and Bardet-Biedl syndrome (BBS). US is the most common inherited cause of combined deafness and blindness while BBS is characterised by rod-cone dystrophy, obesity, polydactyly, learning disabilities and hypogonadism.
Table 1. Estimated percentage and prognosis of RP types.2
| Type of RP | % | Prognosis |
| Sporadic | 30 | Good |
| Autosomal dominant | 25 | Best |
| Autosomal recessive | 20 | Poor |
| X-linked | 15 | Poorest |
| Syndromic RP | 10 | Dependent on the type of underlying syndrome |
History and examination
RP typically presents as complaints of visual disturbances, beginning in childhood or in the early teenage years. However, patients may present in their 40s as this is usually when severe visual impairment occurs. Either way, patients are first assessed by primary care physicians who then make an ophthalmology referral.
History
Patient with suspected RP will primarily complain about not being able to see well in low-light environments or will have difficulty adapting when moving from a well-lit area to a dark area. Patients tend not to notice they have reduced peripheral vision unless the symptoms have been occurring over a prolonged period of time.
Typical symptoms of RP include:
- Reduced peripheral vision or “tunnel vision”
- Nyctalopia (night blindness)
- Impaired dark adaptation
- Photopsia (flashing lights)
- Glare
- Reduced central vision
Rod photoreceptors are affected earlier and more severely than cone photoreceptors in most forms of RP, thus giving rise to the above symptoms.
Other important areas of the history include:
- Past medical history: including previous ocular issues and infections such as syphilis or rubella which can mimic the symptoms of RP.
- Medication history: chloroquine and thioridazine hydrochloride can cause ocular signs similar to RP.
- Family history: it is critical to determine the type of inheritance pattern by drawing a genetic pedigree for the patient’s family, as genetic inheritance is related to the prognosis of RP.
- Occupation and driving: RP may have implications for the patient’s job and their ability to drive.
Clinical examination
A thorough eye and fundoscopy examination should be performed in patients with suspected retinitis pigmentosa. Please see the Geeky Medics guide here for a description of the exam.
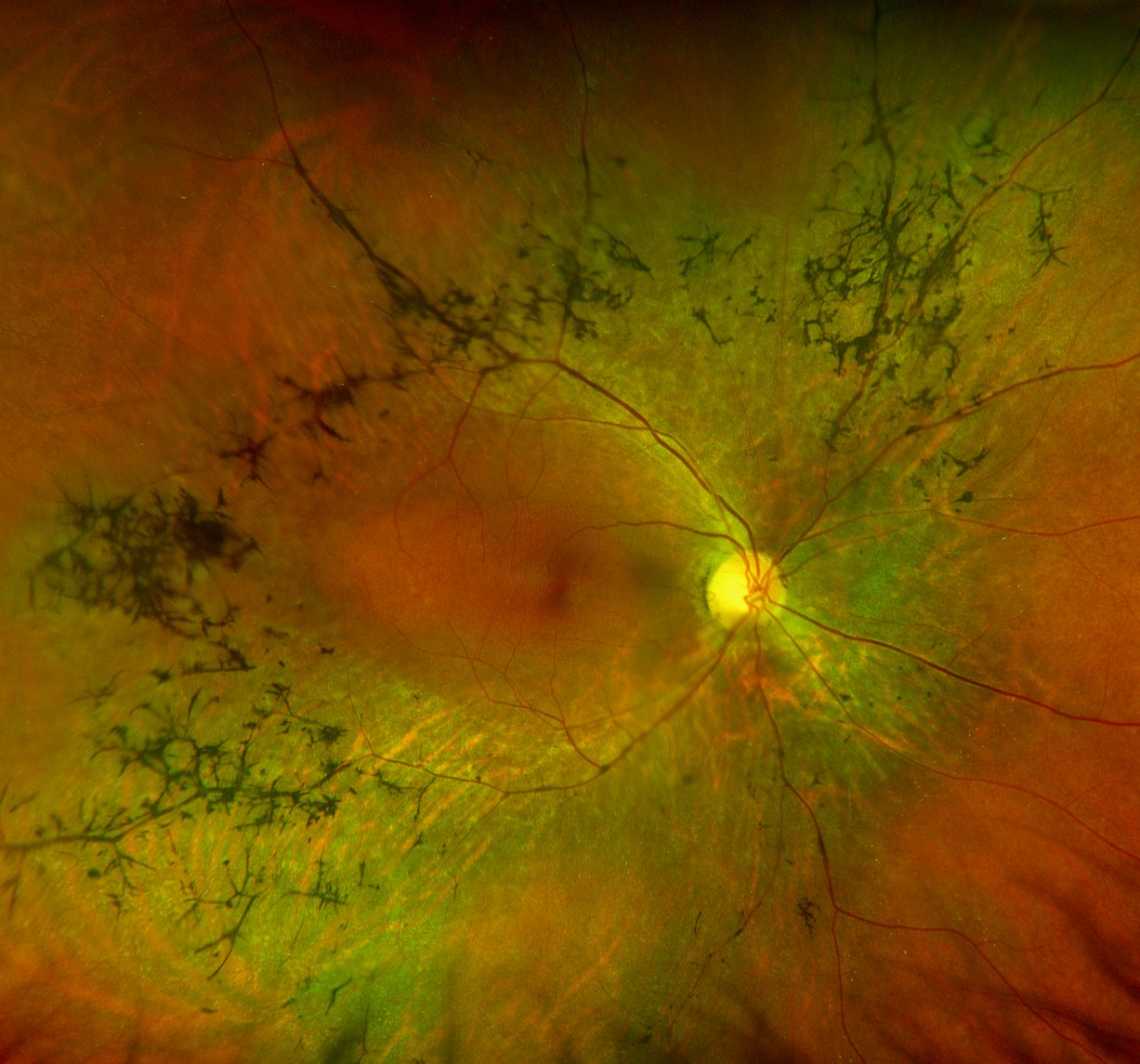
Typical clinical findings on fundoscopy in RP include:
- Bone-spicule retinal pigmentation
- Arteriolar attenuation
- ‘Waxy’ optic disc pallor
- Retinal pigment epithelium (RPE) atrophy
Other findings which may be noted in RP include:
- Myopia
- Cataract (posterior subcapsular subtype)
- Optic disc drusen
- Cystoid macular oedema (CMO)
Differential diagnoses
The clinical presentation of RP is similar to several other conditions that cause pigmentary retinopathy.
Genetic
Genetic diseases which can present with similar clinical features to RP include:
- Leber congenital amaurosis
- Gyrate atrophy
- Choroideremia
Infection
Infections which can present with similar clinical features to RP include:
- Syphilis
- Rubella
- Diffuse unilateral subacute neuroretinitis
Drug toxicity
Drugs which can present with similar clinical features to RP include:
- Chloroquine
- Thioridazine hydrochloride
Investigations
Bedside investigations
Relevant bedside investigations include;
- Assessment of visual acuity (VA) with a Snellen chart: patients may have normal VA or myopia.
- Confrontational visual field assessment: reduced peripheral vision is a typical finding.
- Fundoscopy: typical findings include bone-spicule retinal pigmentation, arteriolar attenuation, ‘waxy’ optic disc pallor and RPE atrophy.
Specialist investigations
Relevant specialist investigations performed by ophthalmology include:
- Electroretinogram: typical findings include a reduction in ‘a’ and ‘b’ wave amplitudes and implicit time may be prolonged in early disease. Patients with advanced disease may have a non-detectable ERG.
- Perimetry (formally assesses any visual field defect): a characteristic ring scotoma in the mid-periphery of the visual field is most commonly noted.
- Genetic testing: to assess for rhodopsin gene mutation and other common variants.
- Optical coherence tomography: to confirm the presence of cystoid macular oedema (CMO).
Management
Conservative
Conservative management involves supportive measures to optimise vision including:
- Low vision aids (e.g. glasses, magnifiers)
- Visual rehabilitation
- Sunglasses (to limit UV exposure)
Medical
Medical management involves treating RP associated complications such as CMO with carbonic anhydrase inhibitors (e.g. topical dorzolamide initially or oral acetazolamide if no improvement).4
Surgical
Surgical management involves treating RP associated complications such as cataracts.
Follow-up
Patients with RP typically receive annual follow-up to detect treatable vision-threatening complications and provide support.
Complications
Complications of RP include:
- Cataracts
- Cystoid macular oedema (CMO)
Prognosis
There is no cure for RP and treatments are only modestly effective in slowing down retinal degeneration.
Patients typically lose 50% of their remaining visual field every 5 years, however complete visual loss is rare.4
Key points
- RP is the most common hereditary retinal dystrophy, resulting in retinal degeneration and progressive vision loss.
- Most cases of RP result from sporadic mutations of the rhodopsin gene.
- Typical symptoms of RP include reduced peripheral and night vision.
- The most common clinical findings in RP include the classic clinical triad of bone-spicule retinal pigmentary changes, arteriolar attenuation and ‘waxy’ disc pallor.
- Conservative management involves the use of low vision aids, visual rehabilitation and sunglasses.
- Medical management involves the administration of oral or topical carbonic anhydrase inhibitors for secondary CMO.
- Surgical management involves cataract surgery.
- Complications of RP include cataract and CMO.
References
- Fight for Sight. Retinitis pigmentosa. Published in 2019. Available from: [LINK]
- Ferrari et al. Retinitis pigmentosa: genes and disease mechanisms. Published in 2011. Available from: [LINK]
- Denniston AKO, Murray PI. Oxford Handbook of Ophthalmology. Published in 2018.
- Grover S, Fishman GA, Anderson RJ et al. Rate of visual field loss in retinitis pigmentosa. Published in 1997. Available from: [LINK]
- Image kindly provided by Dr Peng Yong Sim, Ophthalmology Registrar.
Reviewer
Dr Peng Yong Sim
Ophthalmology Registrar
Editor
Hannah Thomas
