

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Thalassaemia is a group of genetic disorders that lead to reduced haemoglobin in red blood cells. They are classified according to the globin chain which is affected and the severity of the resulting clinical picture, which ranges from asymptomatic to fatal.
The most severe forms of thalassaemia are rare in the UK, but their incidence is rising due to migration.1
Epidemiology
Genetic disorders of haemoglobin are the most common genetic disorders worldwide. Globally 80-90 million people (1.5% of the global population) are affected by thalassaemia.2
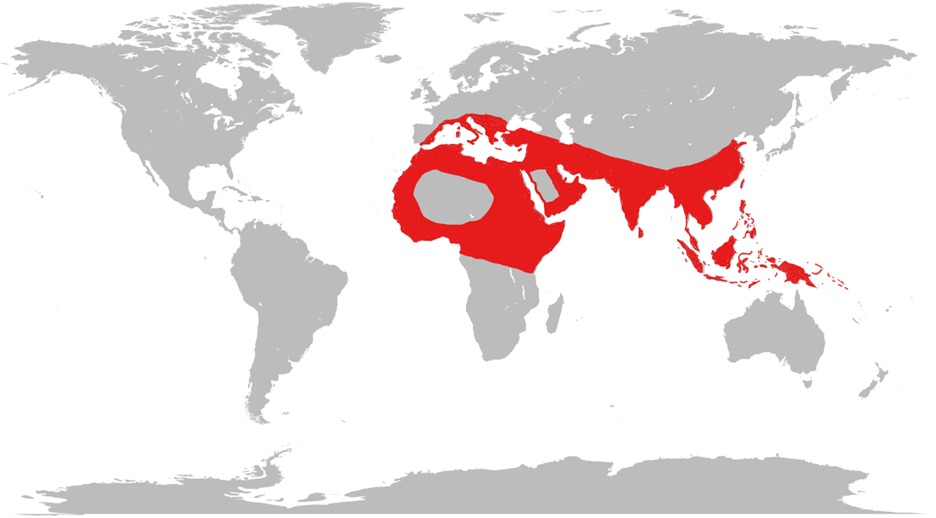
Beta thalassaemia is common in countries from the Mediterranean down to Africa and across to the Middle East and South East Asia.3 Alpha thalassaemia is common in a similar distribution, most commonly found in West Africa and South East Asia.
This geographical distribution is thought to result from the thalassaemia trait offering resistance to the severest forms of malaria, the so-called ‘malaria hypothesis‘. However, this mechanism is not fully established.4
Migration is causing thalassaemia to become more common in other regions, particularly Northern Europe and North America.3
Aetiology
Anatomy
To understand thalassaemia, we first need to understand the structure of haemoglobin.
Haemoglobin is a substance found in red blood cells which carries oxygen from the lungs to the body’s tissues.
One haemoglobin molecule consists of four globin chains (chains of protein forming a roughly spherical shape) connected by noncovalent bonds. These form a structure called a tetramer (one which is made up of four subunits). Each globin chain is associated with a haem group containing an iron molecule.
The most abundant haemoglobin in normal adults is HbA. This is made up of two globin chains, known as alpha chains, and two known as beta chains.6-8
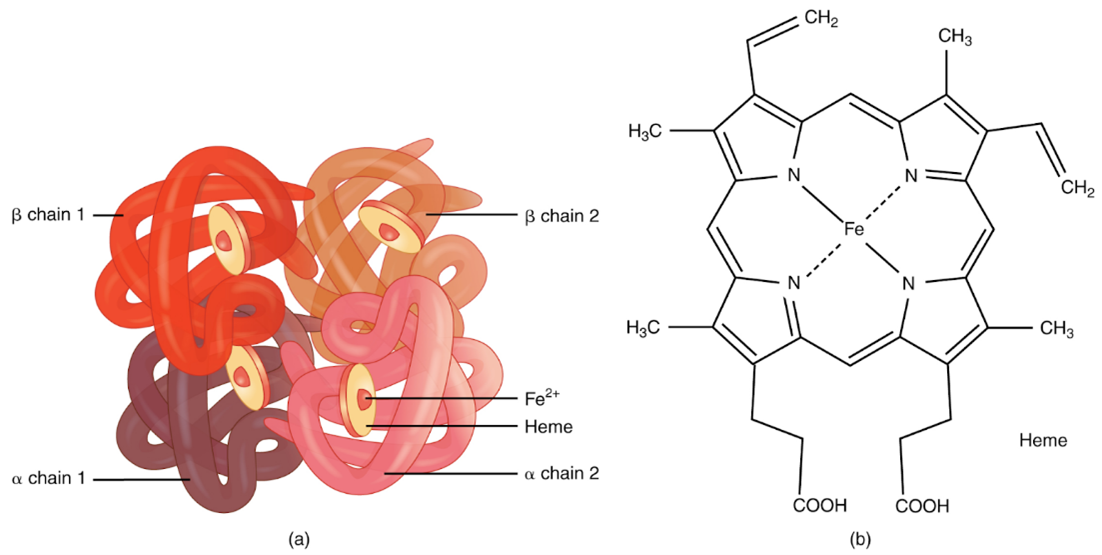
In humans, there are four alpha globin genes located on chromosome 16 used to make alpha chains. There are two beta globin genes located on chromosome 11 used to make beta chains.6-8
Pathophysiology
In thalassaemia, globin genes are absent or mutated, limiting the ability to produce the globin chains encoded by the affected genes.
With reduced production in one type of globin chain, excess numbers of the other type of globin chain accumulate within red blood cell precursors. The accumulation of unpaired globin chains disrupts the cells’ normal function and causes them to become unstable.
These unstable red blood cell precursors cannot mature correctly and are destroyed by macrophages in the bone marrow. The body compensates by producing more red blood cells, but each one has a reduced amount of haemoglobin (leading to hypochromic red cells).
In severe forms of thalassaemia, the deficit of haemoglobin results in insufficient oxygen delivery to the tissues. The most severe forms of thalassaemia have a greater degree of anaemia, in some cases so severe that they are fatal.7,8
Classification
The two main types of thalassaemia are named according to the globin chain whose production is reduced:
- Alpha thalassaemia occurs when there is reduced synthesis of alpha globin chains due to affected alpha globin genes.
- Beta thalassaemia occurs when there is reduced synthesis of beta globin chains due to affected beta globin genes.
Alpha thalassaemia will cause an excess of unpaired beta globin chains, and beta thalassaemia will cause an excess of unpaired alpha globin chains.
Clinical severity
Thalassaemia is also classified depending on the clinical severity, which usually correlates with the number of genes affected.
People with thalassaemia trait (also known as thalassaemia minor) produce a reduced amount of globin chains and thus have increased numbers of small red blood cells, but are asymptomatic with mild/no anaemia seen in their full blood count.
Patients with thalassaemia major produce very little or no relevant globin chains, leading to profound anaemia.
Patients with thalassaemia intermedia have an intermediate clinical picture.10
Alpha thalassaemia
Alpha thalassaemia is caused by deletions of alpha globin genes located on chromosome 16. The clinical severity of the condition is proportional to the number of the four alpha globin genes affected.
Alpha thalassaemia trait/minor
In alpha thalassaemia trait, one or two genes are affected. This is associated with mild anaemia (Hb >100 g/L) and microcytic hypochromic red cells on peripheral blood film.
Alpha thalassaemia trait is asymptomatic. Haemoglobin electrophoresis or high-performance liquid chromatography (HPLC) is normal. These tests measure the relative amounts of different haemoglobin using their differing electrical charges.
In alpha thalassaemia trait, the percentage of each haemoglobin is unchanged because all the haemoglobins found in the normal adult (HbA, foetal haemoglobin/HbF and HbA2) contain alpha chains, so the production of each of them is affected to the same extent.
Alpha thalassaemia trait can be mistaken for iron deficiency anaemia. Accurate documentation of alpha thalassaemia trait is essential to avoid unnecessary investigations and treatment.
When two genes are affected, but they are not on the same copy of chromosome 16, the person has alpha plus thalassaemia. When both alpha globin genes on the same copy of chromosome 16 are affected, the person has alpha zero thalassaemia.
This is important because if two carriers of alpha thalassaemia zero produce a child, there is a one in four chance that the resulting foetus will have alpha thalassaemia major and have no alpha globin genes, a condition which is fatal in utero from hydrops fetalis (see below). 10

Haemoglobin H disease
When three alpha globin genes are affected, this is known as haemoglobin H disease due to the formation of haemoglobin H (a tetrameter of beta globin chains).
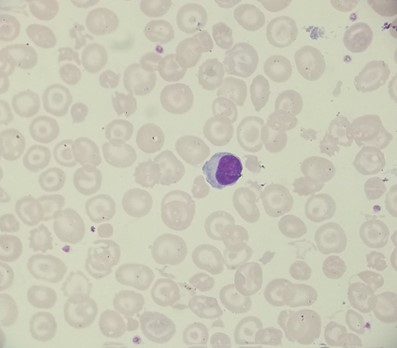
Patients with HbH disease have moderate anaemia and microcytic, hypochromic red cells on a blood film along with anisopoikilocytosis (red blood cells of varying and abnormal sizes and shapes), teardrop cells and basophilic stippling.
Patients with HbH disease may experience symptoms of anaemia or those related to increased red cell turnover (e.g. pigment gallstones or splenomegaly). On haemoglobin electrophoresis or HPLC, a reduced amount of HbA will be seen alongside large amounts of HbH.
Patients should receive folic acid supplementation due to the high turnover of red cells, and subsequent erythropoiesis, depleting folate stores.
The severity of HbH disease varies. Some patients with HbH will need regular transfusions and iron chelation therapy, whilst others may be largely asymptomatic.10
Alpha thalassaemia major (hydrops fetalis)
When four alpha globin genes are affected, there is no alpha chain synthesis. No functional haemoglobin can be produced beyond the embryonic stage of development, and severe anaemia ensues. This is incompatible with life and leads to death in utero by hydrops fetalis (heart failure of the foetus).
On haemoglobin electrophoresis or HPLC, no HbA or HbF is seen. Instead, only Hb Barts (tetramers of gamma chains which in health would pair up with alpha chains to make HbF) are seen.6,7,10
Summary of alpha thalassaemia
Table 1. Summary table of alpha thalassaemia.7,10
| Condition | No. of affected alpha genes | Clinical features | Electrophoresis |
|
Unaffected |
0 |
No symptoms |
Normal |
|
Alpha thalassaemia minor/trait |
1 or 2 |
Usually asymptomatic |
Normal |
|
Haemoglobin H disease |
3 |
Mild-moderate anaemia |
|
|
Alpha thalassaemia major (hydrops fetalis) |
4 |
Death in utero |
|
Beta thalassaemia
Beta thalassaemia is caused by deletions or mutations of the beta globin gene on chromosome 11.
Beta thalassaemia is divided broadly into two groups of diseases depending on the number of genes affected:
- If one copy of the beta globin gene is affected it is referred to as thalassaemia trait/minor
- If both copies of the beta globin gene are affected, the condition is beta thalassaemia major (or beta thalassaemia intermedia if there is some function of the beta gene and some beta chains are still produced, resulting in a less clinically severe condition)
Beta thalassaemia trait/minor
In beta thalassaemia trait/minor, only one copy of the beta globin gene is affected, resulting in a minimal deficiency in beta chains. Beta thalassaemia trait is asymptomatic. This is because the other copy of the beta globin gene can compensate for the missing copy.
The full blood count will show microcytosis with mild or no anaemia. Investigations in these patients include a blood film, showing hypochromic, microcytic red blood cells and target cells. Haemoglobin electrophoresis or HPLC shows a raised level of haemoglobin A2 (HbA2) which is a haemoglobin consisting of two alpha and two delta chains, which is present in small amounts in the normal adult.
As seen in alpha thalassaemia trait, the implications of a diagnosis of this condition are to prevent misdiagnosis of iron deficiency anaemia and to inform prenatal counselling.6,7,10
Beta thalassaemia major
In beta thalassaemia major, both beta globin genes are affected, leading to very few or no beta chains being produced.
Clinical features
Clinical features of beta thalassaemia major include:
- Severe anaemia: becomes apparent at 3-6 months after birth when the switch from foetal haemoglobin (HbF) to adult haemoglobin (HbA) occurs. If the diagnosis has not been made at birth, and a transfusion programme started, the infant will present with typical features of anaemia (e.g. failure to thrive, pallor)
- Splenomegaly and hepatomegaly: occur due to excessive red blood cell destruction and extramedullary (outside the bone marrow) red blood cell production in an attempt to correct the anaemia. In infants, this presents as a swollen abdomen.
- Bone expansion: the bone marrow (where red blood cells are produced) expands into the cortical bone. This occurs because the bone marrow increases in size to produce more red blood cells in response to anaemia.
- Iron overload: occurs due to regular blood transfusions used to treat thalassaemia and enhanced absorption of dietary iron in response to anaemia-induced tissue hypoxia. If iron overload is left untreated, it can lead to organ failure as iron accumulates in various parts of the body (e.g. liver, pancreas, bones and heart).6,12
Features of bone marrow expansion
Bone marrow expansion is prevented by initiating a blood transfusion programme in infancy, but historically presented with the following:
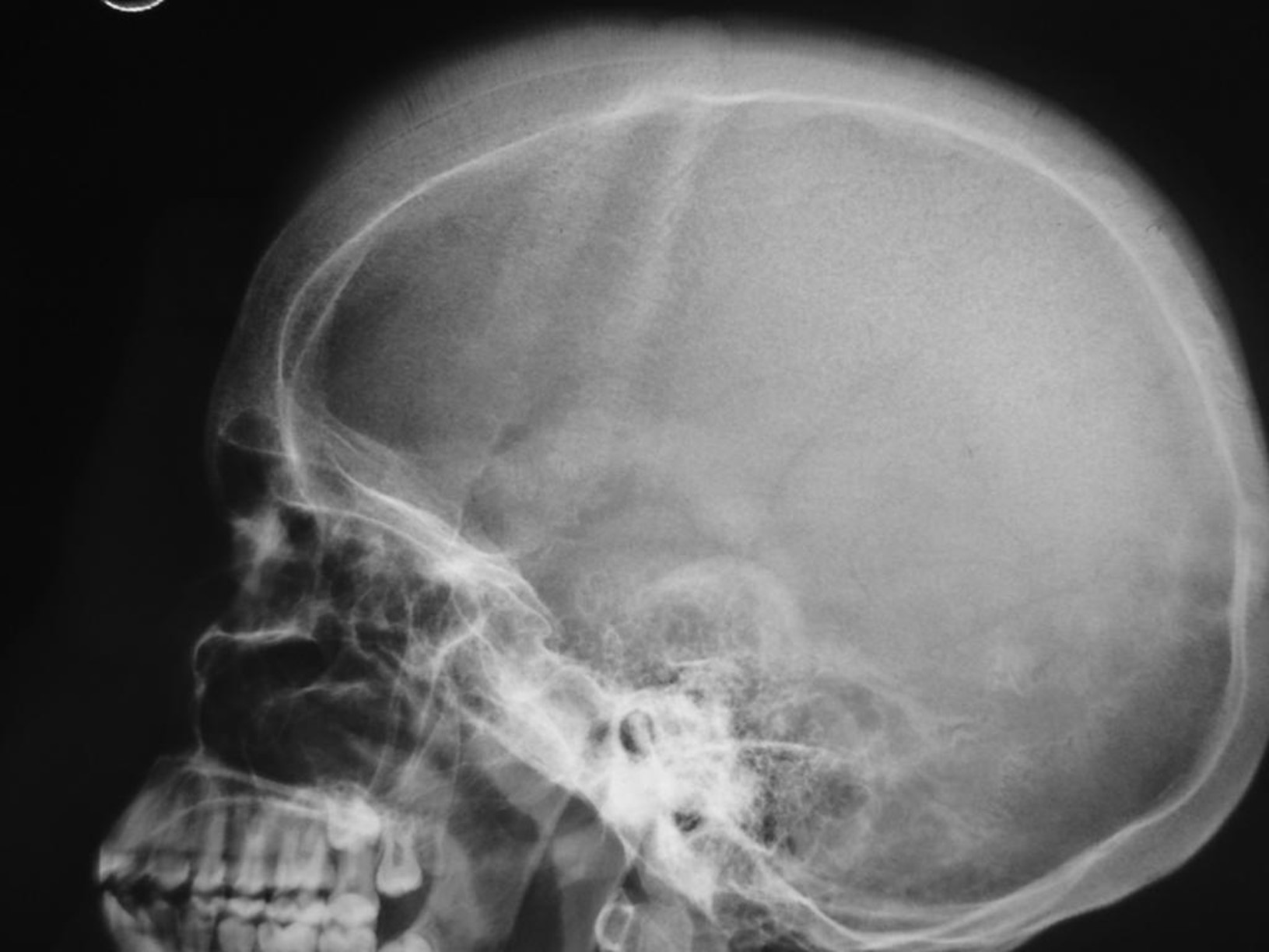
- Thalassaemic facies: occurred when bone expansion affected cheekbones and forehead
- Predisposition to fractures: due to an impairment of bone structural integrity
- Bossing (overgrowth) of the skull with a ‘hair on end’ appearance on X-ray (Figure 5)
Investigations
Most people in the UK will have thalassaemia major detected at or before birth with antenatal or newborn screening, so these investigations are usually not required to make the diagnosis.
Relevant investigations include:6,12
- Full blood count: microcytic anaemia
- Liver function tests: isolated hyperbilirubinemia (the rest of the liver function tests are normal)
- Ferritin or iron studies: normal iron levels, differentiating thalassaemia from iron deficiency anaemia
- Blood film: hypochromic, microcytic anaemia, nucleated red blood cells, target cells, polychromasia (reticulocytes) and basophilic stippling
- Haemoglobin electrophoresis or HPLC: absence of HbA, both HbF and HbA2 are elevated
Management
Management of beta thalassaemia major includes:
- Regular red cell transfusions. Patients usually require 2-3 units every 4-6 weeks to maintain haemoglobin levels over 100g/L and suppress erythropoiesis (the production of red blood cells).
- Splenectomy can be considered to reduce blood requirements. If performed, then appropriate vaccinations and antibiotics need to be given.
- Monitoring of iron status and iron chelation therapy (e.g. desferrioxamine) is essential to avoid iron overload. Iron chelation has side effects, including the potential damage to the liver, kidneys, vision, hearing and bones. Patients require careful monitoring by a thalassaemia specialist.13
- Folic acid (5mg daily) to replace folate stores which are used up more quickly because of increased red cell turnover.
- Regular monitoring of other organ systems (e.g. thyroid function, pulmonary function, cardiovascular function, bone density). Viral serology is also undertaken regularly for HIV, hepatitis B and hepatitis C due to the high number of blood transfusions these patients need.
Future therapies for beta thalassaemia major
Allogeneic stem cell transplantation offers the prospect of a permanent cure with the replacement of red blood cell precursors. However, it is associated with a high risk of morbidity or mortality.
Gene therapies, which involve adding functional genes to stem cells, are also currently in clinical trials.6,8,12
Beta thalassaemia intermedia
In beta thalassaemia intermedia, both beta globin genes are mutated, but the body can still make some functional HbA.
This clinical diagnosis is between the asymptomatic beta thalassaemia trait and the transfusion-dependent beta thalassaemia major. These patients should be offered folic acid supplements. They may require occasional transfusions during increased physiological stress (pregnancy, surgery).15
Differential diagnoses
Whilst thalassaemia major is unmistakable, differential diagnoses of thalassaemia trait include:10,12
- Iron deficiency anaemia
- Anaemia of chronic disease
Screening
Both alpha and beta thalassemia is inherited in an autosomal recessive fashion. However, there are twice as many alpha genes as beta genes.
If both parents have beta thalassemia trait (only one gene), the following outcomes for the baby exist:
- One in four chance of not having the disease and not carrying the gene
- Two in four chance (i.e. one in two) of having beta thalassaemia trait
- One in four chance of having beta thalassaemia major
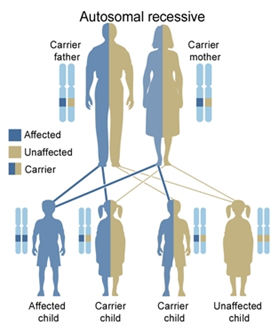
Babies with severe disease can arise from parents who are both silent carriers of thalassaemia.
The NHS has a screening programme for all pregnant women in the UK. Pregnant women are offered a blood test before they reach 10 weeks gestation to identify carriers of thalassaemia.
If the mother is found to be a carrier, the father is offered testing. If both parents are found to be carriers, they are offered a diagnostic test (chorionic villus sampling or amniocentesis) to establish the baby’s thalassaemia status.
Testing for thalassaemia is also included in the newborn blood spot for babies 5-8 days old. 16
Complications
The majority of thalassaemia complications are transfusion-related. Complications include:
- Infection: hyposplenism increases the risk of pneumococcal, Haemophilus and meningococcal infection. Iron overload predisposes to bacterial infections such as klebsiella, yersinia and fungal infections. Viruses can also be very rarely transmitted via blood transfusion.
- Osteoporosis: presents earlier than the general population due to thalassaemia or iron overload. Vitamin D and calcium can be used to prevent osteoporosis.
- Transfusion reactions: including febrile reaction, acute haemolytic reaction and transfusion-related acute lung injury (TRALI).
- Alloimmunisation: with repeated transfusions, there is also the risk of alloimmunisation, where the transfused patient develops antibodies against red cell antigens, which can lead to difficulties in crossmatching blood and transfusion reactions.
- Endocrine complications: due to iron overload, patients can develop delayed sexual maturation, infertility, thyroid dysfunction and parathyroid dysfunction.
- Cardiovascular complications: iron overload increase the risk of arrhythmias and heart failure due to iron-induced cardiomyopathy.
- Thrombosis: thalassaemia increases the risk of thrombosis.
- Other complications: gallstones, leg ulcers, gout and skin bronzing due to iron overload.10-12
Prognosis
This varies depending on the severity of thalassaemia. Thalassaemia trait does not affect life expectancy.
Beta thalassaemia major (and transfusion-dependent HbH disease) are life-limiting conditions, with most patients dying in their 50s and 60s. However, the long-term outlook improves dramatically when iron overload is managed with good chelation therapy. The leading cause of death in these diseases remains heart failure, secondary to iron-induced cardiomyopathy.6,10,12
Key points
- Thalassaemia is a group of conditions which affect the quantity of haemoglobin in red blood cells.
- Thalassaemia is most common in West Africa and South East Asia and affects around 1.5% of the global population.
- Thalassaemia is caused by deletions or mutations in the genes that code for globin chains that make up haemoglobin in red blood cells
- There are different types of thalassaemia, with varying levels of clinical severity depending on how many globin genes are affected
- The UK has a screening programme to identify thalassaemia trait in pregnant women and their partners and thalassaemia major in newborn babies.
- This varies depending on the severity of thalassaemia, thalassaemia trait does not affect life expectancy however beta thalassaemia major is a life-limiting condition.
Reviewer
Dr Angela Collins
Consultant Haematologist
Editor
Dr Chris Jefferies
References
- Hickman M, Modell B, Greengross P, Chapman C, Layton M, Falconer S, et al. Mapping the prevalence of sickle cell and beta thalassaemia in England: estimating and validating ethnic-specific rates. Pubmed. Published in 1999; 104(4):860-7.
- Kattamis A, Forni GL, Aydinok Y, Viprakasit V. Changing patterns in the epidemiology of β-thalassemia. Pubmed Published in 2020; 105(6):692-703
- Galanello R, Raffaella O. Beta-thalassaemia. Orphanet Journal of Rare Diseases. Published in 2010. 5,11.
- Azvolinsky A, Malaria and Thalassemia in the Mediterranean Basin. ASH Clinical News. Published in 2019. Available from: [LINK]
- Crates. Adapted by Geeky Medics. World map blank without borders. License: [CC-BY-2.0]
- Hoffbrand VA and Moss PA. Hoffbrand’s Essential Haematology. Published in 2015.
- Redhouse white, Vanbergen, Crash Course Haematology and Immunology, Fifth Edition. Published in 2019
- Tidy C, Thalassaemia- Patient UK. Published in 2018. Available from: [LINK]
- OpenStax College. Hemoglobin. License: [CC-BY-3.0]
- Kwiatkowski JL. BMJ Best Practice. Alpha-thalassaemia. BMJ Best Practice Available from: [LINK]
- SpicyMilkBoy. Haemoglobin H disease. License: [CC-BY-4.0]
- Sheth S. BMJ Best Practice. Beta-thalassaemia. Available from: [LINK]
- BNF. Desferrioxamine mesylate. Available from: [LINK]
- Dr Iqbal Naseem. Radiopaedia. Hair on end appearance. License: [CC BY-NC-SA]
- Asadov C, Alimirzoeva Z, Mammadova T, Aliyeva G, Gafarova S, Mammadov J. β-Thalassemia intermedia: a comprehensive overview and novel approaches. PubMed. Published in 2018; 108(1):5-21
- NHS. Screening for sickle cell and thalassaemia. Available from: [LINK]
- GenesInLife. Autosomal recessive inheritance. License: [CC-BY-3.0]
