

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Haemophilia is a largely inherited bleeding disorder of variable severity.
There are two main types: haemophilia A, caused by a clotting factor VIII deficiency, and haemophilia B (also referred to as Christmas disease) which is caused by a factor IX deficiency. The deficiency can be quantitative or qualitative, meaning the factor can be inadequate in terms of quantity or function.
The severity of both diseases is generally dependent on the levels of factor VIII or IX. Laboratory factor levels are reported as a percentage of normal factor activity, with the normal range between 50 and 150%. This can also be converted into international units, where 100% factor activity is equal to 1.00 U/ml.
Roughly half of haemophilia patients are classed as having severe disease, with factor levels below 1%. The remaining haemophilia patients are classed as either moderate, with factor levels of 1-5%, or mild, with levels from 5 to 50%.
In 2019, the UK prevalence for haemophilia A was approximately 1 in 8000, whilst the prevalence for haemophilia B was roughly 1 in 35000.1
Aetiology
Haemophilia A is caused by mutations in the factor VIII gene, whilst haemophilia B is caused by mutations in the factor IX gene. Both genes are mapped to the X chromosome and are passed down in a recessive fashion, so males with a single mutation will have the disease, whilst females, with two X chromosomes, will be carriers for the disease.
In the rare case of a female having a homozygous mutated factor gene, inherited from an affected father and a carrier mother, it has been observed that they can present with haemophilia symptoms.2
Occasionally, haemophilia can be acquired due to the formation of autoantibodies targeting factor VIII or IX, or as a result of malignancy.3
Haemostasis physiology
Haemostasis is the process of stopping bleeding and preventing further blood loss from a damaged blood vessel. It is a three-step process, triggered by damage to the blood vessel endothelium:
- Blood vessel vasoconstriction
- Primary haemostasis
- Secondary haemostasis
After the blood vessel constricts, which decreases pressure downstream, circulating platelets come into contact with the collagen of the broken vessel and become activated. Activated platelets start to plug the hole, and also release ADP and thromboxane A2, two chemicals that can activate other platelets in a positive feedback mechanism. This unstable and fragile platelet plug is named the primary plug.
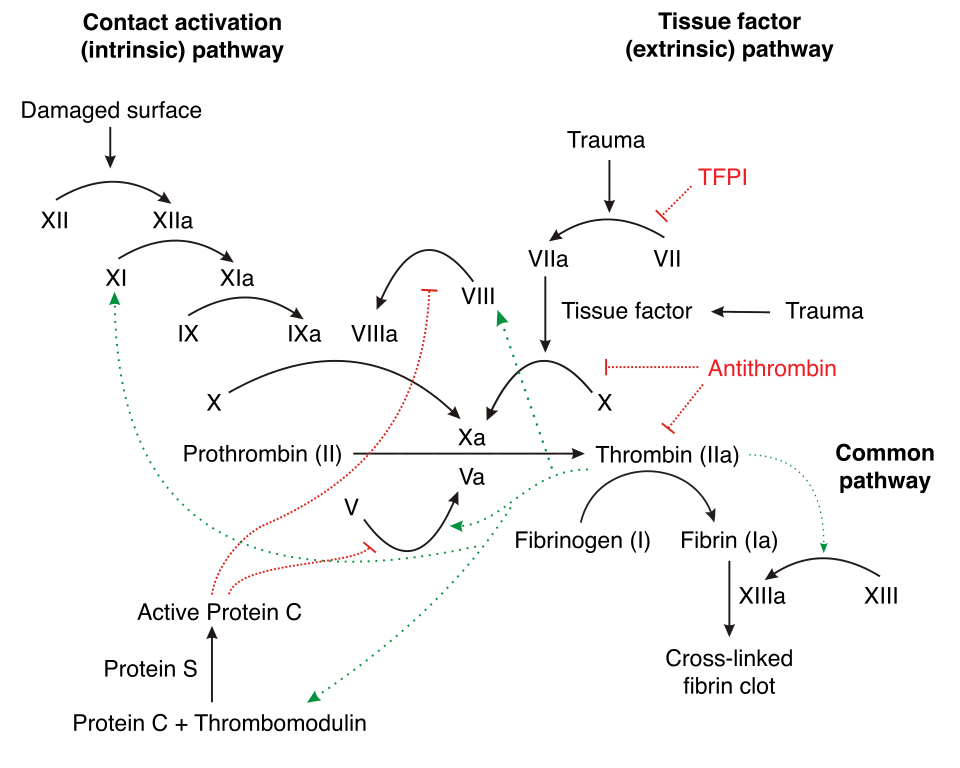
When clotting factors make contact with the sub-endothelium or extravascular components, the clotting cascade is triggered. The clotting cascade is a series of reactions whereby factor zymogens become activated and, in turn, activate the next zymogen in the chain.
There are two arms of this amplifying factor cascade, the intrinsic and extrinsic cascades, which both lead to activation of factor X. Activated factor X turns prothrombin into thrombin, which activates fibrinogen into fibrin (in addition to further stimulating the coagulation cascade). Fibrin filaments accumulate around and stabilise the primary platelet plug, forming the secondary plug.
Both factor VIII and IX are involved in the intrinsic cascade, which is initially triggered by the activation of thrombin and factor XII by platelets surrounding the vascular injury. If either one of these factors is deficient, this cascade works ineffectively, slowing the rate of secondary plug formation and risking blood loss through the fragile primary plug.
Clinical features
Haemophilia can present differently depending on patient age and disease severity.
Typically, inherited cases present at younger ages and acquired cases occur in older individuals. More severe disease, characterised by lower factor levels, often presents earlier in life (even as early as during delivery).
On the other hand, mild haemophilia patients often do not realise they have the disease, and only present after a bleed following major trauma or surgery, or precipitated by drugs such as non-steroidal anti-inflammatories (NSAIDs) or aspirin.
History
Severe haemophilia will typically present early with spontaneous or prolonged bleeding.5
Typical symptoms of haemophilia may include:
- Severe epistaxis
- Bleeding gums
- Haematuria: gross or microscopic (i.e. detected on dipstick).
- Intra-articular or intramuscular bleeds: commonly affected joints include the knees, ankles and elbows.
- Excessive bruising/ecchymoses, contusions or spontaneous haemorrhage during childhood play.
- Prolonged bleeding after a surgical or dental procedure, or post-venepuncture.
Other less common bleeding presentations include:
- Intracranial haemorrhage: presenting with neurological symptoms.
- Haematemesis, melaena and frank rectal bleeding (from gastrointestinal bleeding).
- Haemoptysis
- Compartment syndrome
When taking a history, other important areas to cover include:
- Secondary symptoms such as fatigue or frequent infections, as they may indicate alternative diagnoses (e.g. haematological malignancy).
- Drug history: several different medications can alter the ability of the clotting cascade to function effectively (e.g. warfarin, aspirin, NOACs).
- Social history: consider the possibility of violence as the cause of excessive bruising or bleeding (e.g. domestic violence).
- Family history: given that most haemophilia is inherited, it’s essential that you take a comprehensive family history.
Clinical examination
Clinical examination may reveal signs suggestive of haemophilia such as bruising, haematoma, active bleeding and joint swelling (haemarthrosis).
Differential diagnoses
The haemostatic and fibrinolytic systems within the body normally counteract each other to maintain balance. Several medical conditions can result in loss of this balance, resulting in excessive bleeding.
Common differential diagnoses for haemophilia include:
- von Willebrand’s disease (VWD): especially subtype 2N
- vitamin K deficiency (vitamin K is crucial for the synthesis of many clotting factors)
- disorders of the fibrinolytic system
- hepatic disease (many clotting factors, as well as several anticoagulative proteins, are synthesised in the liver)
- deficiencies of specific clotting factors
Platelet disorders can also present similarly to haemophilia, although platelet deficiencies typically cause petechial haemorrhages and ecchymoses (bruising) rather than the haematomas and haemarthroses which are more characteristic of clotting factor deficiencies.
Iatrogenic causes of spontaneous or prolonged bleeding are also a possibility in patients being treated with anticoagulants or antiplatelets.
Investigations
Full blood count and extended clotting screen
It is important to carry out a full blood count (FBC) and extended clotting screen in any patient presenting with unexplained spontaneous or prolonged bleeding symptoms. This can help diagnose haemophilia and rule out many of the differential diagnoses.
Findings associated with haemophilia include:
- Reduced haemoglobin and low haematocrit on FBC: can indicate a recent or chronic bleed.
- Normal platelet count on FBC
- Normal prothrombin time (PT), bleeding time (BT), fibrinogen levels and von Willebrand factor levels.
- Prolonged activated partial thromboplastin time (APTT): although this can be normal in mild disease.
- Reduced factor VIII or factor IX activity level: for haemophilia A or B respectively.
PT measures the extrinsic and common pathways. APTT measures the intrinsic and common pathways. Both factor VIII and IX form part of the intrinsic clotting pathway, so deficiencies affect the APTT reading.
For more information, see the Geeky Medics guide on interpreting a coagulation screen.
Mixing study
A mixing study is another useful investigation of haemophilia. By mixing a haemophilia patient’s blood plasma in a 1:1 ratio with normal plasma, a prolonged APTT should normalise. This is because the normal plasma contains functional clotting factor, so the deficiency can be bypassed.
Imaging
Imaging can be carried out in emergency situations to detect bleeds or in the chronic setting to identify signs of degenerative joint disease. Suitable modalities used to diagnose bleeds include CT, MRI and Doppler ultrasound.
Management
Prophylaxis
Prophylactic treatment is given in order to prevent disease, in this case bleeding events. Prophylaxis also helps to preserve long-term joint function by reducing episodes of haemarthrosis.
Severe forms of haemophilia can require prophylactic factor infusions, replacing the missing or non-functional clotting factor, in order to prevent frequent haemarthroses or other bleeding episodes. Factor infusions should ideally be administered until the child reaches physical maturity, but are often continued long-term, under the continuous review of a haematologist.
The genetically engineered factor injections are given as a slow intravenous bolus and in some cases may be required up to three times a week. These doses should be tailored to the patient, such as administration just prior to a child’s physical education lessons.
Patients with severe haemophilia are at high risk of bleeding during and after surgery. For this reason, their factor activity should be increased to 50-100% for 2-7 days before surgery, and closer to 100% for surgery on the brain or prostate. Tranexamic acid can be considered as this inhibits fibrinolysis without increasing thrombosis risk in healthy individuals.
Monitoring of factor and factor inhibitor levels (see complications) should occur regularly during prophylaxis, and the Haemophilia Joint Health Score (HJHS) should be calculated frequently. Additionally, patients should wear a medical emergency identification bracelet stating the disease, normal factor level and other important information.
Patients should also avoid competitive contact sports and unnecessary manual labour, as these increase the risk of haemarthroses and head injuries. Other exercises, including racquet sports or swimming, should be encouraged.
To protect against bloodborne diseases, all patients and carers that may inject blood products should be vaccinated against hepatitis A and hepatitis B. In haemophilia patients, these immunisations should be given subcutaneously rather than intramuscularly to avoid bleeding and haematoma formation.
Acute bleeds
For any bleeding episodes, normal physical methods of bleeding cessation should be advised. Depending on the severity of bleeding, patients may need factor infusions, fresh frozen plasma or cryoprecipitate.
For major haemorrhage, including those involving the central nervous, gastrointestinal and genitourinary systems the aim is to correct factor levels to 100%.
For minor haemorrhages, such as haemarthrosis, oral mucosal and intramuscular bleeds, the aim is to increase factor levels to 30-50%. These raised factor levels should be maintained through further infusions for several days.
If possible, coagulation tests and a group and save sample should be taken prior to treatment, however, these investigations should not delay treatment.
Haematomas and haemarthroses can be very painful and require analgesic treatment. The oral route is preferred, although NSAIDs should be avoided as they increase the risk of gastrointestinal bleeding. Intramuscular opiates should also be avoided as this mode of delivery can result in intramuscular bleeding and haematoma formation.
It is possible to treat acute bleeds in mild haemophilia A patients with desmopressin (DDAVP), which stimulates von Willebrand factor (vWF), which in turn promotes factor VIII activity.
Calculating factor dose
There are formulas published to calculate the dose required to treat a bleed in a haemophilia A or B patient.6
Doses are rounded up to the nearest vial size, as slight overtreatment has minimal detrimental effect. They are as follows:
Haemophilia A dose formula: weight (kg) * factor level increase desired (%) * 0.5 = number of factor VIII units needed
Haemophilia B dose formula: weight (kg) * factor level increase desired (%) = number of factor IX units needed
NB: Always check factor product literature as this may vary.
Genetic counselling and pregnancy
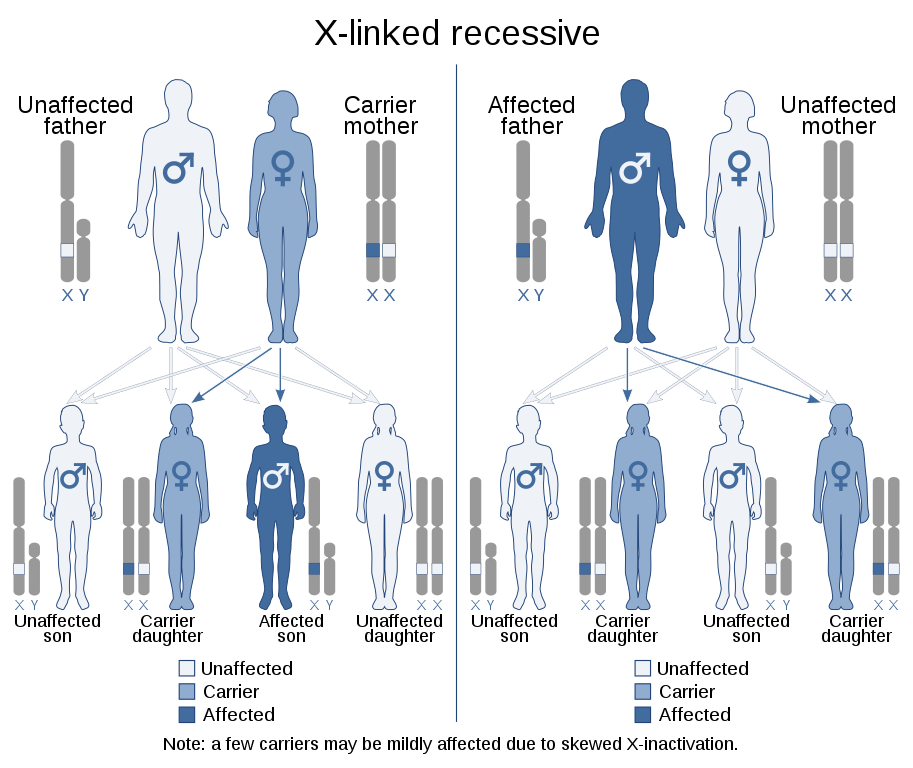
As haemophilia has an X-linked recessive inheritance pattern, a daughter of a male haemophilia patient will be a carrier whilst a son will be unaffected, as they inherit their father’s normal Y chromosome.
There is a 50% chance that a daughter of a female carrier will be a carrier and a 50% chance that a son will be affected. This information should be discussed with any haemophilia patients or carriers looking to have a child.
Genetic testing and counselling should be made available to prospective parents. During pregnancy, the fetus can be tested for a haemophilia mutation via:
- Chorionic villus sampling at 11-14 weeks: removing a small portion of the placenta for testing.
- Amniocentesis at 15-20 weeks: sampling the amniotic fluid for testing.
Both procedures carry small risks of premature labour and miscarriage, which should be assessed by the family prior to the procedure. The obstetric haematology team should be involved in the management of pregnancy and birth.
A haemophilia diagnosis should be made as soon as possible after delivery, by testing of uncontaminated cord blood. Recombinant factor should be administered immediately after a positive haemophilia diagnosis.
Complications
If untreated, severe disease, and repeated bleeding into joints, can lead to arthropathy (painful and reduced range of movement), joint deformity, soft tissue haemorrhages, retroperitoneal bleeds or haematoma formation. These can present as surgical emergencies and may require operative intervention.
Bloodborne viruses
Before 1985, factor concentrate was plasma-derived and created from multiple blood donations. As a result, many haemophilia patients were infected with bloodborne viruses, including HIV, hepatitis B and hepatitis C. The prevalence of plasma-derived factor viral infection is higher in haemophilia A patients, as the condition is more common, was more often severe (requiring factor treatment) and more plasma from multiple donors was needed for their concentrate.
One UK-based survey carried out in 1988 recorded 59% of severe haemophilia A and 11% of severe haemophilia B patients tested positive for HIV antibody.8
Many haemophilia patients to this day are currently infected with HIV and hepatitis viruses, which together produce a worse prognosis, and lead to other disease states, including liver failure and hepatocellular carcinoma. This is not a concern for newly diagnosed patients, as this risk has been virtually eliminated with the invention of safer and more efficacious recombinant factor products.
Antibody inhibitor formation
Antibody inhibitor formation affects nearly a third of haemophilia A patients, as well as a smaller proportion of haemophilia B patients, reducing the efficacy of factor therapy. Inhibitors, or factor antibodies, form as the patient’s immune system recognises therapeutic factor as non-self and forms an immune response against these proteins.
The immune response can result in anaphylaxis on exposure to any factor product, so first exposure should always take place in a specialist centre.5
It is more common for inhibitors to diminish the effect of factor treatment, a state that worsens over time and so should be monitored. One method of treatment is immune toleration induction (ITI), which involves administering small quantities of factor regularly over a period of time until inhibitor antibody levels decrease.
This treatment is usually offered to severe haemophilia A patients but is avoided in severe haemophilia B patients, as it is often less effective and carries a risk of anaphylaxis. Immunosuppressants can also be offered to haemophilia patients with mild disease to avoid inhibitor formation.
Key points
- Haemophilia is a bleeding disorder, most often inherited in an X-linked recessive fashion.
- Factor VIII or IX deficiency is responsible for a phenotype involving spontaneous and prolonged bleeding.
- APTT is often prolonged, and reduced factor activity levels can be used to diagnose haemophilia.
- Recombinant factor infusions can be given prophylactically to prevent bleeds and joint deformity or therapeutically to treat acute bleeds.
Reviewer
Dr Richard Gooding
Consultant haematologist
Editor
Dr Chris Jefferies
References
- United Kingdom Haemophilia Centre Doctors’ Organisation. UKHCDO Annual Report 2019 including Bleeding Disorder Statistics for 2018/2019. Published in 2019. Available from: [LINK]
- Nair P.S., Shetty S. and Ghosh K.. A Homozygous Female Hemophilia A. Published in 2012. Available from: [LINK]
- Franchini M., Lippi G.. Acquired Factor VIII Inhibitors. Published in 2008. Available from: [LINK]
- Joe D. Coagulation with arrows for negative and positive feedback. Licence: [CC-BY-SA]. Available from: [LINK]
- NHS.uk. Haemophilia. Published in 2020. Available from: [LINK]
- Hemophilia of Georgia. Calculating the Dose. Available from: [LINK]
- SUM1. X-linked recessive inheritance scenarios for either the mother being a carrier or the father being affected. Licence: [CC-BY-SA]. Available from: [LINK]
- AIDS Group of the United Kingdom Haemophilia Centre Directors. Prevalence of antibody to HIV in haemophiliacs in the United Kingdom: a second survey. Published in 1988. Available from: [LINK]
