

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Retinoblastoma is the commonest intraocular malignancy in infancy affecting 1 in 20,000 live births.1 The retinal nerve cells grow uncontrollably, resulting in a retinoblastoma.
Aetiology
Hereditary retinoblastoma is due to an inherited mutation in the RB1 gene on chromosome 13. RB1 is a tumour suppressor gene, and hereditary retinoblastoma often affects both eyes.
Sporadic retinoblastoma is due to a new mutation in the RB1 gene in one of the child’s retinal cells and often affects one eye.
Risk factors
The following risk factors are associated with an increased likelihood of retinoblastoma:
- Family history of retinoblastoma
- Known genetic mutation of the RB1 gene
- Previous retinoblastoma diagnosis
Clinical features
Typical clinical features of retinoblastoma include:2
- Unilateral or bilateral vision loss
- Strabismus
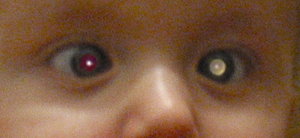
- Leukocoria (white pupil appearance)
- Bulging of one eye (unilateral retinoblastoma)
- Red and painful eye
In the context of retinoblastoma, a thorough ophthalmic examination is required by an ophthalmologist. A dilated eye examination and a red reflex examination are performed to look at the retina of the eyes. The loss of red reflex may be identified on the NIPE.
Differential diagnoses
Differential diagnoses to consider in the context of suspected retinoblastoma include:
- Cataracts
- Retinopathy of prematurity
- Vitreous haemorrhage
- Other retinal tumours (such as astrocytic hamartoma)
Investigations
Laboratory investigations
Relevant laboratory investigations include:
- Baseline blood tests (FBC, U&E, LFTs): to assess general fitness and exclude other conditions
- Genetic testing: for RB1 gene mutation (associated with hereditary retinoblastoma)
Imaging
Relevant imagining investigations include:
- Fundus photography: allows the tumour appearance and location to be visualised
- Fluorescein angiography: helps identify abnormal blood vessels and fluid build-up surrounding the tumour
- Ophthalmic ultrasound: to identify the tumour size and composition
- MRI scan: can check for any abnormalities of the pineal gland and are better than CT at showing the structures of the eye and brain
Diagnosis
The gold standard diagnostic test for retinoblastoma is ultrasound imaging.3 Clinical examination of the eye and MRI scans are useful to confirm the diagnosis. Unlike most other cancer types, retinoblastoma is not diagnosed with a biopsy when the tumour is confined to the eyes alone.
Management
Medical management
Larger tumours, or patients with metastatic disease, require chemotherapy (using agents such as carboplatin and vincristine). Smaller tumours can be treated with local therapies, such as cryotherapy and laser therapy.
Radiotherapy is used in patients with unsuccessful medical management.
Surgical management
Surgical management of retinoblastoma involves an operation to remove the eye (enucleation). This is performed as a last resort when the tumour has advanced, and the vision has been lost. Subsequently, an artificial ocular prosthesis is fitted.4
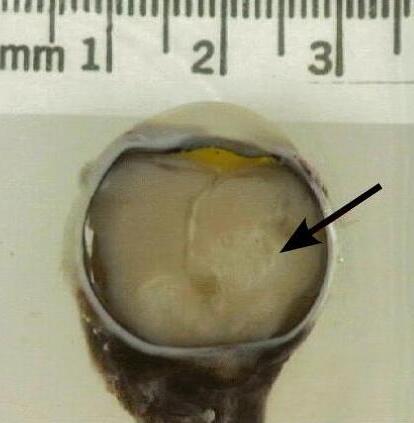
Complications
Complications of retinoblastoma include:
- Retinal detachment
- Retinal necrosis
- Optic nerve invasion
- Blindness
- Cataracts
- Subsequent malignant neoplasms
The complications of retinoblastoma management include:6
- Sensorineural hearing loss from chemotherapy
- Retinoblastoma recurrence
- Retinal detachment post cryotherapy
- Retinopathy post radiation
Prognosis
The prognosis of retinoblastoma is favourable, with a 5-year relative survival rate of 97% for children with retinoblastoma. Death from retinoblastoma is rare and primarily arises from the spread of tumours via the meninges or optic nerve. Metastases can travel further through the circulation to other viscera and skeletal structures.
Key points
- Retinoblastoma is the most common intraocular cancer in children
- Hereditary retinoblastoma (which typically affects both eyes) occurs due to an inherited mutation in the RB1 gene
- Clinical examination of the eye reveals a white pupil known as leukocoria, and there will be loss of the red reflex
- Occular ultrasound helps assess the tumour size and composition and is the gold standard diagnostic test for retinoblastoma
- Enucleation of the eye is often a last resort for managing retinoblastoma when other treatments have not worked
- The prognosis is good, with a 5-year relative survival rate of 97% for children with retinoblastoma
Reviewer
Dr Omer Jamall
ST5 Ophthalmology
Editor
Dr Chris Jefferies
References
- Black, G. C., Ashworth, J. L., & Sergouniotis, P. I. (Eds.). (2022). Clinical ophthalmic genetics and genomics. Academic Press.
- J Morley-Smith. A child with a white eye reflection as a result of retinoblastoma. License: [Public domain]
- Moulin, A. P., Gaillard, M. C., Balmer, A., & Munier, F. L. (2012). Ultrasound biomicroscopy evaluation of anterior extension in retinoblastoma: a clinicopathological study. British journal of ophthalmology, 96(3), 337-340.
- Ancona-Lezama, D., Dalvin, L. A., & Shields, C. L. (2020). Modern treatment of retinoblastoma: A 2020 review. Indian journal of ophthalmology, 68(11), 2356.
- US Government. Gross pathology of retinoblastoma tumor in enucleated eye of three year old female. License: [Public domain]
- StatPearls. Retinoblastoma. Published in 2023. Available from: [LINK].
