

- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Multiple sclerosis (MS) is an autoimmune inflammatory disease of the central nervous system (CNS) which is characterised by demyelination. It is the most common progressive neurological disorder in high-income countries, and in young adults.1,2
Aetiology
Pathophysiology
MS is an autoimmune inflammatory process in the central nervous system thought to likely be mediated by aberrant T-cell activation.3
The underlying cause of this process, however, remains unclear.2-5
It is thought to be a complex relationship between a genetic predisposition and exposure to environmental triggers.2,4
Classification
It is helpful to think of MS as a continuous spectrum of disease, where the rate of progression and severity worsens over time.
However, for management and research purposes it is simpler to categorise patients into four main groups.
Clinically/radiologically isolated syndrome (CIS/RIS)
Clinically isolated syndrome (CIS) is an otherwise unexplained clinical episode of neurologic dysfunction, and radiologically isolated syndrome (RIS) is evidence of white matter pathology on neuroimaging not attributable to any other pathology in the absence of clinical symptoms.4
Both syndromes have features suggestive of MS, however, neither CIS nor RIS satisfies the diagnostic criteria that must be met to make a diagnosis of MS (see McDonald’s criteria in investigations section).3-7
Patients who present with a CIS/RIS are much more likely than the general population to develop MS. Some 34% of patients with RIS develop acute neuropathic symptoms consistent with multiple sclerosis within 5 years.4
Relapsing-remitting MS (RRMS)
Relapsing-remitting MS (RRMS) is by far the most common form of disease at presentation, encompassing approximately 85% of patients.3,5,6
RRMS constitutes unpredictable attacks of neurological dysfunction (lasting >24 hours in the absence of fever), followed by relief of symptoms though patients may not return fully to baseline function.3-7
Secondary progressive MS
Secondary progressive MS initially presents as RRMS, then later declines steadily and progressively without remission.3-7
Primary progressive MS
Primary progressive MS is a steady, progressive worsening of disease severity from the onset without remission.3-7
Risk factors
Risk factors for MS include:1-4
- Family history
- Sex (F>M)
- Age between 25-35
- Other co-existing autoimmune diseases, such as type 1 diabetes mellitus
- Smoking
- Previous EBV infection; infectious mononucleosis / raised anti-EBV nuclear antigen 1 (EBNA1) antibody titres
- Latitude of habitat
- Vitamin D deficiency
Clinical features
History
While there are no clinical findings that are unique to MS, there is a range of recognised characteristic symptoms (table 1).
The most common symptoms on initial presentation are:5
- Limb numbness/tingling
- Limb weakness (subacute onset)
- Cerebellar symptoms
Certain phenomena are also considered characteristic of MS:5
- Uhthoff’s phenomenon: worsening of symptoms on exercise/in warm environments (e.g. in a bath).
- Lhermitte’s phenomenon: lightning-shock pain down the spine on flexion of the neck secondary to cervical cord plaque formation.
Table 1. An overview of the clinical features of MS.
| System affected | Clinical features |
|
Central |
Motor:
Sensory:
Cerebellar:
Fatigue Depression/labile mood |
|
Ophthalmic |
Nystagmus Optic neuritis (pain on movement, visual field defect, loss of colour vision – particularly red) Diplopia – internuclear ophthalmoplegia (INO) |
|
Otolaryngeal |
Dysphagia Slurring/stuttering speech |
|
Muscular |
Weakness Cramping Spasm/contractures |
|
Urinary |
Urinary frequency Incontinence Retention |
|
Gastrointestinal |
Constipation/diarrhoea |
Other important areas to cover in the history include:
- Past medical history (such as any history of focal neurologic deficit, or other autoimmune diseases)
- Family history of MS/other autoimmune diseases
- Smoking status
- Diet
- Impact on activities of daily living
- Driving status
- Falls risk assessment
Clinical examination
MS has the potential to involve multiple different systems. As such, it is important to carry out a thorough neurological examination including a cranial nerve examination and cerebellar examination.
Patients with MS may have a wide range of clinical signs on cranial nerve examination.
Optic nerve (CN II)
Optic neuritis, which is usually monocular, can be the first sign of MS. Fundoscopy may reveal blurring of the optic disc in the acute setting, though often no changes are apparent. A previous episode of optic neuritis is often characterised by disc pallor, which is often far more helpful.
Relative afferent pupillary defect (RAPD) is another manifestation MS which may be revealed by the pendular swinging light test.
The pupil of the healthy eye will constrict as light is shone on it, exhibiting a normal direct light reflex. The contralateral pupil will also constrict as there is a normal consensual light reflex.
When the light is then swung to the affected eye, the previously constricted pupil will dilate as there is no afferent (outgoing) signal being transmitted by the inflamed optic nerve, impairing the direct light reflex.
See the Geeky Medics visual assessment guide for more information on pupillary reflexes.
Oculomotor (CN III), trochlear (CN IV) and abducens (CNVI) nerve
Ophthalmoplegia arises from involvement of the cranial nerve nuclei, resulting in symptoms of a cranial nerve palsy, or involvement of the medial longitudinal fasciculus (MLF) resulting in internuclear ophthalmoplegia (INO).
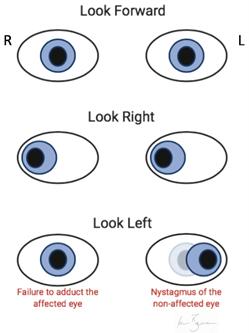
INO is a conjugate lateral gaze palsy, where there is a failure of adduction of the affected eye during horizontal eye movement (figure 1).
Nystagmus is usually observed with the abduction of the contralateral eye, as it tries to normalise the two discordant images being sent to the brain simultaneously (figure 1).
Trigeminal nerve (CN V)
Involvement of CN V nucleus may result in facial paraesthesia, and/or weakness of the muscles of mastication.
Facial nerve (CN VII)
CN VII lesions will result in weakness of facial muscles of expression. This can mimic the signs of an acute stroke. MS is an upper motor neuron lesion, and as such there will be sparing of frontalis controlling expression of the forehead.
Vestibulocochlear nerve (CN VIII)
Loss of balance, and/or sensorineural deafness.
Glossopharyngeal (CN IX), vagus (CN X) and hypoglossal nerve (CN XII)
Loss of motor function to tongue/pharynx resulting in speech and swallowing problems.
Accessory nerve (CN XI)
Loss of motor function to sternocleidomastoid and trapezius resulting in neck weakness and hypertonia.
Cerebellar signs
MS has the potential to affect any part of the CNS. As such, sometimes patients may present with symptoms of cerebellar dysfunction including:
- Nystagmus: slow, large-amplitude nystagmus while eyes are resting at the midline, flickering towards the side of the lesion.
- Intention tremor: a slow, coarse tremor that gets worse on the extension of a limb towards an intended object/target, often associated with dysmetria.
- Scanning dysarthria: sentences or even words are broken up into a number of separate syllables that can be expressed at varying volume. It may appear as though the patient is searching for (or scanning for) the correct next word/sound.
Peripheral nervous system signs
Sensory signs:
- Abnormal sensation
- Romberg’s test positive: this suggests the involvement of the dorsal column of the spinal cord, affecting proprioception.
Motor signs:
- Hypertonia
- Decreased power
- Hyperreflexia
- Clonus
Differential diagnoses
The presentation of multiple sclerosis can be varied. MS can resemble a broad number of other disorders which should be considered/ruled out ahead of making a diagnosis:8
- Migraine with aura
- Hypoglycaemia
- Hypothyroidism
- Electrolyte abnormalities (e.g. hyponatraemia)
- Peripheral neuropathy (e.g. B12 deficiency, diabetic neuropathy, radiculopathy, motor neuron disease)
- Stroke
- Space occupying lesion (e.g. glioblastoma/meningioma/lymphoma or cerebral abscess)
- Compression of brainstem or spinal cord (e.g. Chiari malformation, cervical spondylosis, disc herniation)
- Infection (e.g. Lyme disease, syphilis, HIV)
- Inflammatory/autoimmune (e.g. sarcoidosis, systemic lupus erythematosus, CNS vasculitis)
Investigations
Laboratory investigations
Laboratory investigations are important for ruling out other causes of neurological dysfunction:8,9
- Full blood count: white cell count for infection.
- C‑reactive protein: a marker of an inflammatory process (e.g. infective/autoimmune).
- Liver function tests: for basic baseline biochemistry and to rule out hepatic pathology associated with MS mimics.
- Urea and electrolytes: to rule out electrolyte disturbance, which might mimic MS.
- Calcium and angiotensin-converting enzyme: to rule out sarcoidosis.
- Thyroid function tests: to rule out hypothyroidism.
- Vitamin B12: to rule out B12 peripheral neuropathy.
- HIV serology: to rule out HIV.
Radiological investigations
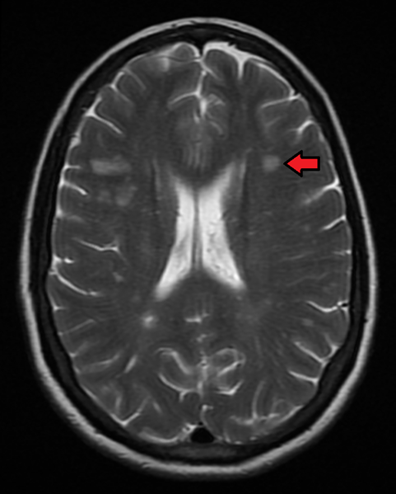
The main investigation for MS is MRI of the brain and spinal cord with gadolinium contrast. MS lesions will be apparent as T2-hyperintense white matter plaques (figure 2).
Other investigations
Lumbar puncture for CSF analysis is useful, especially when there is no clear radiological evidence of white matter pathology consistent with MS on MRI (see McDonald’s criteria below).
Typical findings in the CSF of an MS patient include a high protein content and oligoclonal bands of immunoglobulin aggregates on CSF electrophoresis.9
CSF electrophoresis must be accompanied by serum protein electrophoresis (SPEP) concurrently for results to have any meaning. Oligoclonal bands should not appear on SPEP.
Evoked potentials are also a supportive investigation that historically would have been considered useful in the workup of MS, visual evoked potentials being the most useful. In clinical practice today it is considered solely supportive and not diagnostic in isolation.10
McDonald diagnostic criteria
The McDonald criteria outline the clinical, radiological and biochemical findings which must be met to make a diagnosis of MS.6
It is centred around the principle that MS is a disease which demonstrates pathology that is disseminated in time (i.e. events occur at >1 distinct time periods) and disseminated in space (i.e. ≥2 lesions affecting distinctly different locations in the CNS).
Table 2 describes further what dissemination in time and space means in a clinical and radiological context, and table 3 describes how the McDonald criteria apply this information to make a diagnosis of MS.
Table 2a: Description of the clinical findings required to satisfy the McDonald criteria, focussing on the necessity to demonstrate dissemination in time and dissemination in space.
| Clinical findings | |
|
Dissemination in time For example, 2 episodes of left leg weakness that have occurred months apart |
Dissemination in space For example, a patient presents with optic neuritis and symptoms of wrist drop |
Table 2b: Description of the radiological findings required to satisfy the McDonald criteria, focussing on the necessity to demonstrate dissemination in time and dissemination in space.
| MRI evidence | |
|
Dissemination in time For example, a patient’s scan demonstrates T2-hyperintense paraventricular white matter lesions, and a second scan 4 months later demonstrates a new juxtacortical white matter lesion that was not present previously ≥2 lesions identified simultaneously in areas of the CNS typically associated with MS*, but where only one lesion enhances upon injection of gadolinium contrast** |
Dissemination in space For example, a patient’s MRI shows evidence of T2-hyperintense white matter lesions in the cerebellum as well as in the spinal cord |
*periventricular, cortical, juxtacortical, infratentorial and the spinal cord
**active lesions will enhance with gadolinium, old/inactive lesions will not
Table 3: The McDonald diagnostic criteria for the diagnosis of MS, adapted from Thompson et al., 2018.6
| Clinical findings | Diagnostic evidence required |
|
Can demonstrate ≥2 neuropathic symptoms that are disseminated in both time and space |
None, clinical evidence sufficient MRI desirable to support diagnosis, but must be consistent with MS |
|
Can demonstrate recurrence of a single neuropathic symptom (i.e. dissemination in time, but not in space) |
Dissemination in space, demonstrated by MRI or await further clinical attack implicating different site |
|
First/single attack that has resulted in ≥2 neuropathic symptoms (i.e. evidence of dissemination in space but not time) |
Dissemination in time, demonstrated by MRI/second clinical attack or presence of oligoclonal bands in the CSF, in the absence of otherwise atypical CSF findings (biochemical evidence of MS) |
|
First attack that has resulted in a solitary neuropathic symptom (i.e. no evidence of dissemination in either space or time) |
Dissemination in space, demonstrated by MRI/second clinical attack an involving a different part of the CNS and dissemination in time demonstrated by MRI/second clinical attack or presence of oligoclonal bands in the CSF, in the absence of otherwise atypical CSF findings (biochemical evidence of MS)
|
Management
Management of an acute episode
NICE guidelines state that a relapse/attack can be diagnosed if a patient presents with:9
- New symptoms or worsening of existing symptoms
- Subacute onset (>24hrs)
- Absence of fever/signs of active infection
Not all relapses require medical intervention. If left alone, some symptoms may remit on their own.
If symptoms are severe, however, or interfere with activities of daily living then you should consider medical intervention. Treatment of an acute attack/relapse requires high dose steroid therapy with methylprednisolone (500mg by mouth for five days, or 1g by mouth for three to five days).4,9
Patients can be managed in the community by their GP or may require admission to hospital for specialist input, depending on the severity of their symptoms and the impact of symptoms on their activities of daily living.9
Plasmapheresis is also an option if the exacerbation is refractory to steroids.4,5
Flares of MS symptoms may also often be caused by something other than an attack, such as an infection. In such an event, treatment of the underlying cause should be the primary concern.
Long-term management
There are currently no curative therapies for MS. Management of the disease focuses on curtailing the demyelination process and managing side-effects of the disease process.
Management of the demyelinating process
Beta-interferon (beta-IFN) and glatiramer acetate are injectable disease-modifying agents regularly used in the management of MS, which have both been shown to be similarly effective in decreasing relapses in RRMS. These medications do not, however, alter disease progression.4,9,11,12
Additionally, there are a number of approved oral disease-modifying therapies for RRMS, including dimethyl fumarate, fingolimod and cladribine.11,12
Monoclonal antibody therapies such as alemtuzumab and natalizumab are effective therapies in the treatment of RRMS. These drugs carry more dangerous side effect profiles. Most notably, natalizumab can rarely cause progressive multifocal leukoencephalopathy via reactivation of the JC virus, a very common virus often otherwise found dormant in glial cells.4,9,11
In addition, alemtuzumab also has had an FDA black box warning for strokes and arterial dissections and thus should be used extremely cautiously.13,14
With regard to PPMS, ocrelizumab is the only medication to date that has been shown to be effective in slowing the progression of symptoms in patients.4,15
It is also worth mentioning the emerging role of autologous haematopoietic stem cell transplantation in patients with MS refractory to disease-modifying drug therapy, which has shown great promise to date.4
The symptoms secondary to the demyelinating process in MS can have marked and severe impacts on a patient’s quality of life. As such, it is paramount that these be treated effectively and in a timely manner. These symptoms are outlined in table 3, along with multidisciplinary management options that should be considered.4,5,9
Other management strategies
Other important management strategies for MS include:
- Amelioration of modifiable risk factors (e.g. smoking cessation, obesity, vitamin D)4,5
- Ensuring immunisations are up to date
A multidisciplinary approach is important, other professionals who may be involved include:
- Nurse specialist involvement for assistance in managing the complex needs individuals with MS, their families and carers experience on a day-to-day basis.
- Physiotherapy for assistance and rehabilitation of balance, strength and mobility difficulties.
- Occupational therapy for management around the home and any modifications/aids required.
- Speech and language therapy for the assessment, diagnosis and rehabilitation of speech and/or swallowing difficulties.
- Social care worker for the management of emotional, social and economic need of patients with MS (e.g. social welfare entitlements).
- Psychologist involvement for any mood disturbance. Conversion disorder and depression are common complications of MS.
- GP involvement in the co-ordination of community care.
Complications
MS is a chronic, progressive disease, with a variable clinical course.3-5
Not much is understood of its aetiology, and as such, there is difficulty identifying reliable biomarkers of disease severity/rate of disability onset.5
Morbidity5
The majority of people diagnosed with MS become unemployed within the following 15 years.
50% of patients require some form of mobility aid within 20 years of diagnosis.
Approximately half of patients eventually develop substantial cognitive deficits.
Mortality5
Patients with multiple sclerosis have a reduction in life expectancy of 7 to 14 years compared with the general population.
At least half die from causes directly related to multiple sclerosis.
Primary progressive disease and older age at onset are associated with shorter survival.
There are numerous recognised complications of MS in addition to the classical motor/sensory symptoms associated with the disease (table 4).
Table 4. An overview of the complications of MS.
|
Complication of MS |
Management |
|
Fatigue |
Physiotherapy input Exercise therapy |
|
Mobility balance problems |
Physiotherapy Occupational therapy, focusing on mobility aids and home modifications Fampridine can be used to increase walking speed in certain patients |
|
Spasticity |
Physiotherapy Consider pharmacological management with baclofen (an anti-spasmodic medication) and/or botulinum toxin injection of the affected muscle(s) |
|
Bowel and bladder dysfunction |
Catheterisation (either intermittent or self) Laxatives Anticholinergic medications can be effective in the pharmacological management of trigonal muscle dysfunction in urinary bladder incontinence. |
|
Emotional lability |
May need psychiatric/psychological management |
|
Ophthalmic complications |
Ophthalmology input Consider gabapentin/memantine in managing oscillopsia |
|
Pain |
Pain specialist |
|
Cognition, including memory |
Involvement of dementia specialists Home assistance Occupational therapy input +/- aids |
Key points
- Multiple sclerosis (MS) is an autoimmune inflammatory disease of the central nervous system (CNS) which is characterised by demyelination.
- Risk factors include being young, having a family/personal history of autoimmune disease and previous infection with EBV. There are also a number of recognised preventable risk factors such as vitamin D deficiency, the latitude of habitat and smoking.
- The presentation of MS can be quite varied, and may potentially involve quite a number of systems.
- Diagnosis is made using the McDonald criteria, which encompasses clinical and radiological findings consistent with MS, disseminated in time and space.
- Examination of MS patients includes the full examination of the peripheral nervous system, along with a cerebellar exam and a cranial nerve exam.
- The mainstay for management of an acute attack is high-dose steroid therapy to reduce inflammation, along with the management of any specific complications.
- Long term management requires multidisciplinary input. Treatment focuses on the disease itself (beta-IFN/Glatiramer acetate + monoclonal antibodies) along with the long-term management of side effects.
Reviewer
Neurology Registrar
Editor
Dr Chris Jefferies
References
- Wallin MT, Culpepper WJ, Nichols E, Bhutta ZA, Gebrehiwot TT, Hay SI, et al. Global, regional, and national burden of multiple sclerosis 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Published in 2019. Available from: [LINK]
- Belbasis L, Bellou V, Evangelou E, Ioannidis JPA, Tzoulaki I. Environmental risk factors and multiple sclerosis: An umbrella review of systematic reviews and meta-analyses. Published in 2015. Available from: [LINK]
- Baecher-Allan C, Kaskow BJ, Weiner HL. Multiple Sclerosis: Mechanisms and Immunotherapy. Published in 2018. Available from: [LINK]
- Thompson AJ, Baranzini SE, Geurts J, Hemmer B, Ciccarelli O. Multiple Sclerosis. Published in 2018. Available from: [LINK]
- ClinicalKey Clinical Overview; Multiple sclerosis Last updated: 08 November 2019. Available from: [LINK]
- Thompson AJ, Banwell BL, Barkhof F, Carroll WM, Coetzee T, Comi G, et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Published in 2018. Available from: [LINK]
- Lublin FD, Reingold SC, Cohen JA, Cutter GR, Sørensen PS, Thompson AJ, et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Published in 2014. Available from: [LINK]
- Miller DH, Weinshenker BG, Filippi M, Banwell BL, Cohen JA, Freedman MS, et al. Differential diagnosis of suspected multiple sclerosis: a consensus approach. Published in 2008. Available from: [LINK]
- NICE. Recommendations | Multiple sclerosis in adults: management | Guidance. Published in 2014, Last updated: 11 November 2019. Available from: [LINK]
- Walsh P, Kane N, Butler S. The clinical role of evoked potentials. Published in 2005. Available from: [LINK]
- Tramacere I, Del Giovane C, Salanti G, D’amico R, Filippini G. Immunomodulators and immunosuppressants for relapsing-remitting multiple sclerosis: A network meta-analysis. Published in 2015.Available from: [LINK]
- Derfuss T, Mehling M, Papadopoulou A, Bar-Or A, Cohen JA, Kappos L. Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Published in 2020. Available from: [LINK]
- McCall B. Alemtuzumab to be restricted pending review, says EMA Reports of stroke in patients having taken alemtuzumab for multiple sclerosis prompt a safety review by the European Medicines Agency. Published in 2019. Available from: [LINK]
- European Medicines Agency Measures to minimise risk of serious side effects of multiple sclerosis medicine Lemtrada. Published in 2020. Available from: [LINK]
- Montalban X, Hauser SL, Kappos L, Arnold DL, Bar-Or A, Comi G, et al. Ocrelizumab versus Placebo in Primary Progressive Multiple Sclerosis. N Engl J Med. Published in 2017. Available from: [LINK]
- Internuclear Ophthalmoplegia. License: [CC BY-SA]. Available from: [LINK]
- James Heilman, MD. Multiple Sclerosis. License: [CC BY-SA]. Available from: [LINK]
