
- 📖 Geeky Medics OSCE Book
- ⚡ Geeky Medics Bundles
- ✨ 1300+ OSCE Stations
- ✅ OSCE Checklist PDF Booklet
- 🧠 UKMLA AKT Question Bank
- 💊 PSA Question Bank
- 💉 Clinical Skills App
- 🗂️ Flashcard Collections | OSCE, Medicine, Surgery, Anatomy
- 💬 SCA Cases for MRCGP
To be the first to know about our latest videos subscribe to our YouTube channel 🙌
Introduction
Cystic fibrosis (CF) is an inherited disease affecting multiple organs. A genetic mutation results in thickened secretions which commonly leads to recurrent respiratory infections.
CF is the most common inherited disease in Caucasians.1 In the UK there are around 10,600 cases, whereas worldwide there are thought to be around 100,000.2
Aetiology
CF is a genetic disease inherited in an autosomal recessive fashion.
Mutations are found on chromosome 7 in the CF transmembrane conductance regulator (CFTR) gene. The most common mutation found in Caucasians is delta-F508 (DF508).1
Normally, the CFTR allows efflux of chloride and inhibits influx of sodium. Usually this keeps sodium and chloride in the lumen, out of the cells.
In CF, more chloride leaves the cell and more sodium is reabsorbed along with water, due to osmosis. Subsequently, secretions are dehydrated and thickened with increased chloride.3
Risk factors
As CF is an inherited condition, family history of the disease is the only risk factor.1
Clinical features
CF can present at any age (although it is usually found in newborn screening) and severity of symptoms varies.1
CF has effects on multiple systems but most commonly presents with respiratory effects.
Thickened secretions reduce mucociliary clearance from the bronchi and increased salt concentration leads to impaired bacterial defences. Subsequently, bacterial colonisation and lung inflammation increases, which can present as recurrent lower respiratory tract infections and bronchiectasis.1
Thickened secretions in sinuses can also lead to recurrent infection and subsequent nasal polyps due to chronic inflammation.4
The gastrointestinal system is affected in several ways. Commonly, pancreatic insufficiency leads to both exocrine and endocrine dysfunction.
Impaired exocrine function leads to malabsorption of nutrients and fat-soluble vitamins, whereas impaired endocrine function results in cystic fibrosis-related diabetes mellitus (CFRD). Blockage of pancreatic ducts can also result in pancreatitis. CF causes thickened bile leading to obstruction of the bile ducts and gallstones.4
Intestinal obstruction in the newborn, termed meconium ileus, can be the presenting feature in infants.3 Infants can also present with rectal prolapse.1 Adults can develop distal intestinal obstruction syndrome (DIOS) which presents with bloating, abdominal pain and vomiting.4
Infertility is common in boys due to congenital absence or fibrosis of the vas deferens. In girls, thickened mucus in the cervix and irregular menstruation can lead to reduced fertility.1
Chloride is not resorbed from sweat glands leading to excess salt in the sweat. This allows diagnosis of CF by sweat testing.1
History
The history should focus on respiratory symptoms but should also include questions about the less common features of CF:
- Respiratory: frequency of infections, cough, sputum (colour, amount), haemoptysis, breathlessness
- Pancreatic function: failure to thrive, steatorrhoea, thirst, polyuria
- Gastrointestinal: abdominal pain, bloating, vomiting
- Difficulty conceiving and irregular menstruation
Clinical examination
A thorough respiratory examination should be performed. Typical findings on respiratory examination may include:
- General inspection of the environment: sputum pots, oxygen (concentrator or cylinder with a mask or nasal cannulae), nebuliser, inhaler
- General inspection of the patient: younger age, increased work of breathing, cachexia, vascular access (Portacath, PICC line)
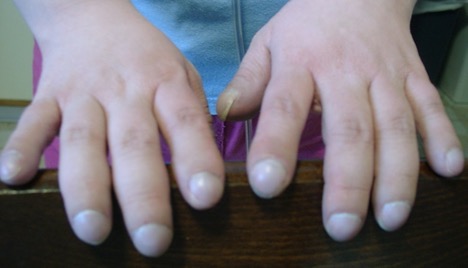
- Peripheral signs: finger clubbing (Figure 1), cyanosis
- Chest signs: coarse crackles over areas of bronchiectasis
Examination for other signs of CF should look for:
- Finger prick marks due to insulin use in diabetes
- Abdominal examination for pain, signs of chronic liver disease, feeding tubes (NG or PEG)
Differential diagnoses
Common pathologies such as pneumonia, malignancy and pulmonary embolism should be considered depending on presenting features. If gastrointestinal features are predominant, causes such as coeliac disease should be considered.
A rare but important differential diagnosis is primary ciliary dyskinesia (PCD). This is an autosomal recessive inherited disease caused by dysfunction of cilia in sinuses, lungs and reproductive organs.4
Investigations
Bedside investigations
Relevant bedside investigations include:
- Urine dip: for glucose in case of diabetes
- Lung function testing: an obstructive picture is most common but can be restrictive, mixed or normal
Laboratory investigations
Relevant laboratory investigations include:1
- Blood tests: FBC and CRP (infection and anaemia), U&Es, fasting glucose, LFTs (biliary or liver effects), vitamin A, C and E levels
- Sputum culture and sensitivity: commonly Haemophilus influenzae, Staphylococcus aureus, Pseudomonas aeruginosa, Burkholderia cepacia
- Sweat testing: high chloride concentration in 98% sensitive
- Genetic testing: for CFTR
- Faecal elastase: for pancreatic insufficiency
Imaging
Relevant imaging investigations include:
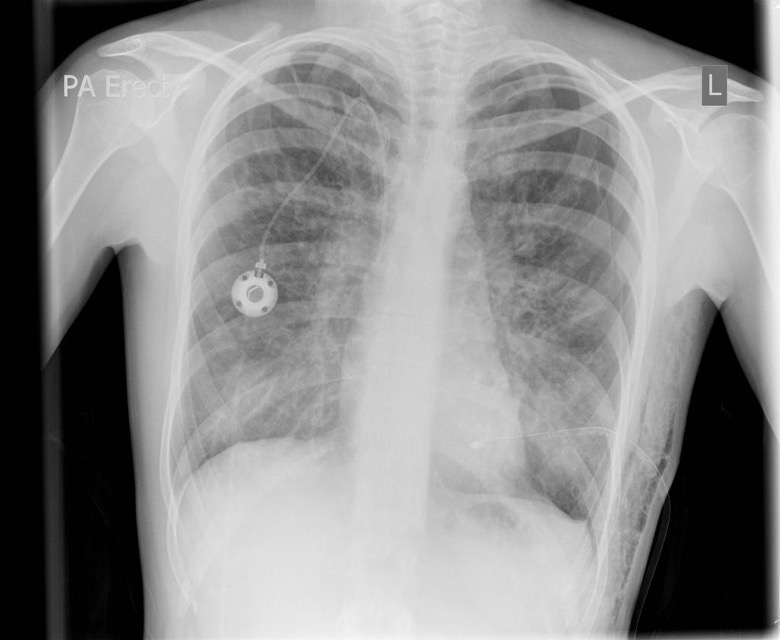
- Chest x-ray: bronchiectasis, hyperinflation
- High-resolution CT chest: bronchial wall thickening, bronchiectasis (tree-in-bud appearance, signet ring sign), mucus plugging (finger in glove)
- CT angiography: in haemoptysis, if endovascular intervention is being considered
Management
CF is usually managed by a multidisciplinary team of specialists in a tertiary centre.1
Medical management
All patients should have annual vaccinations and lifestyle advice for regular physical exercise.
Respiratory manifestations are managed with:
- Chest physiotherapy for mucus clearance with active cycle of breathing
- Prophylactic antibiotics and antibiotics for acute exacerbations guided by sputum culture (oral, inhaled or intravenous)
- Inhaled bronchodilators
- Mucolytics (oral or inhaled): hypertonic saline or dornase alfa
Pancreatic insufficiency is managed with pancreatic enzymes (Creon) and diabetic control (usually with insulin).
Due to recurrent infection and malabsorption, weight should be closely monitored, and nutrition managed by a dietician. Supplementation of fat-soluble vitamins and a high-calorie diet is encouraged. If nutrition cannot be maintained, enteral feeding by PEG or nasogastric tube may be needed.
Fertility counselling should be offered, including information about infertility and the inherent risk in pregnancy.
Follow up
Follow up should be carried out with specialists twice a year, in conjunction with primary care. It should include regular pulmonary function tests, chest x-ray, sputum cultures and diabetes screening.1
Surgical management
The mainstay of surgical management is transplant (lung +/- heart). Complications such as massive haemoptysis or pneumothorax may also be managed surgically.
Complications
Respiratory complications are caused by bronchiectasis and recurrent infections. Progressive airflow obstruction can lead to cor pulmonale and death.
Other respiratory complications include haemoptysis (which can be large volume and life-threatening) and pneumothorax.
Other complications include chronic liver disease (and related sequelae), osteoporosis and psychological effects.
Key points
- Cystic fibrosis (CF) is an autosomal recessive inherited condition due to a mutation in the CFTR gene. The mutation leads to thickened secretions in multiple organs.
- Thickened mucus in the lungs leads to recurrent infections and bronchiectasis, haemoptysis and pneumothoraces.
- Other effects of thickened secretions include nasal polyps, pancreatic insufficiency (malabsorption and cystic fibrosis-related diabetes mellitus), biliary or intestinal obstruction, infertility and excess salt in sweat.
- History and examination should focus on the respiratory system with a brief abdominal examination. Inspection of the environment and patient is important in CF.
- Differential diagnoses include pneumonia, malignancy, pulmonary embolism and rarely primary ciliary dyskinesia.
- Investigations include lung function testing, basic blood tests, sputum culture, sweat test and genetic testing.
- Appropriate imaging for CF includes chest x-ray and high-resolution CT chest which show bronchiectasis.
- Management of CF is multidisciplinary including chest physiotherapy, antibiotics and mucolytics for respiratory manifestations. Nutritional needs should be managed with a high-calorie diet or enteral feeding, vitamin supplementation and pancreatic enzymes.
- Surgical management can include transplant (heart and/or lung) in some cases.
- Respiratory complications include cor pulmonale, haemoptysis and pneumothorax.
Reviewer
Dr Samantha Cockburn
Respiratory Registrar
Editor
Dr Chris Jefferies
References
- Patient info. Cystic Fibrosis. Published in 2020. Available from: [LINK]
- Cystic Fibrosis Trust. Cystic fibrosis FAQs. Available from: [LINK]
- Radiopaedia. Cystic fibrosis (pulmonary manifestations). Published in 2010. Available from: [LINK]
- National Organization for Rare Disorders. Cystic Fibrosis. Published in 2017. Available from: [LINK]
- Radiopaedia/Wikipedia. Finger clubbing in cystic fibrosis. Licence: [CC BY NC SA 3.0]
- Radiopaedia/Dr Jeremy Jones. Bronchiectasis, complicating left sided pneumothorax with chest drain, surgical emphysema, portacath. Licence: [CC BY NC SA 3.0]
